Periodic Table |
 |
 |
 |
 |
 |
 |
| Polyatomic Species: Hybrid & Molecular Orbitals | Functional Group Database |
Organic π-Systems
Organic
chemistry is dominated by the functional group approach. This approach
invokes the experimental observation that ethanal, propanal, butanal, pentanal,
hexanal, etc., all have an "aldehyde", R-CHO, functional group
(FG) and that the spectrum of reactivity of the aldehyde FG is largely independent of the alkyl group to which it is attached. Thus, it is only
necessary to understand the chemistry of the R-CHO aldehyde FG to predict
the chemistry of a whole range of compounds that possess the R-CHO function.
Most functional groups are associated with π-systems,
and this page explores the frontier molecular orbital (FMO) structure of
hydrocarbon π-systems,
and then of the same π-systems
with embedded heteroatoms using Hückel MO theory.
Introduction to Organic Functional Groups
MO theory formally assigns a molecule all encompassing molecular orbitals, however, it is usual and convenient to regard the structure of larger organic molecules as being constructed from discrete functional groups or FGs.
The Hückel principle of σ-π (sigma-pi) separability assumes that as π-electrons are at a much higher energy than the σ-skeleton electrons, the sigma and π-electrons have no influence upon each other. Thus:
| The σ–skeleton of an organic molecule can be described using VSEPR theory, |
- Heteroatoms are embedded into the hydrocarbon sigma skeleton
- Functional groups have characteristic sets of sigma and π FMOs.
The empirical rule is that FGs assume discrete identities – undergo distinct sets of reactions – when separated by two or more alkyl methylene (–CH2–) carbons:
FG–CH2–CH2–FG
Consider phenol, benzyl alcohol, 2-phenyl ethanol and 3-phenyl propanol:
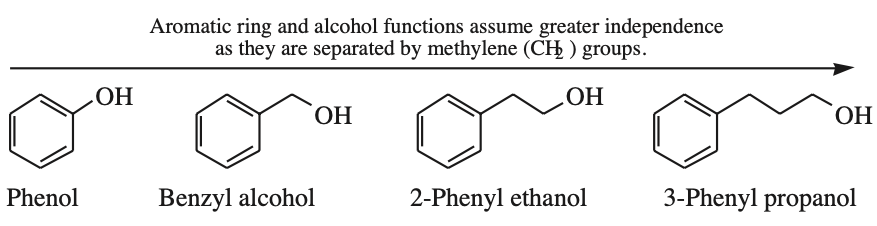
Phenol, Ph-OH, acts as a single function because the alcohol –OH group is directly attached to the aromatic ring
- The hydroxy group has the effect of activating the aromatic ring for electrophilic aromatic substitution
- The aromatic ring renders the hydroxy function a stronger Brønsted acid than an alkyl alcohol.
In benzyl alcohol, PhCH2OH, the aromatic ring has a very small influence upon the alcohol function and vice versa.
With 2-phenyl ethanol, PhCH2CH2OH, the benzene and the alcohol functions have little direct influence (although beta elimination to styrene, PhCH=CH2, is possible.
The aromatic and alcohol functions are effectively independent in 3-phenyl propanol, Ph(CH2)3OH.
Test your knowledge of functional groups using the Chemistry Tutorials & Drills website.
Hückel MO and VB Resonance Construction of Polyene Ribbons π-System
FGs can be modelled by both Hückel molecular orbital (HMO) theory and VB resonance models.
Hückel MO theory is the more sophisticated technique. It can provide quantitative information about orbital phase, electron energy and electron density for linear, branched, cyclic, polycyclic, charged and uncharged, hydrocarbon and heteroatom containing π-systems.
VB resonance theory is more qualitative. Resonance structures are inter converted by curly arrows as an aid to predicting where partial charges (which can be equated with reactive sites) will occur. The VB technique is particularly useful for heteroatom containing π-systems.
The Hückel and VB approaches are complementary.
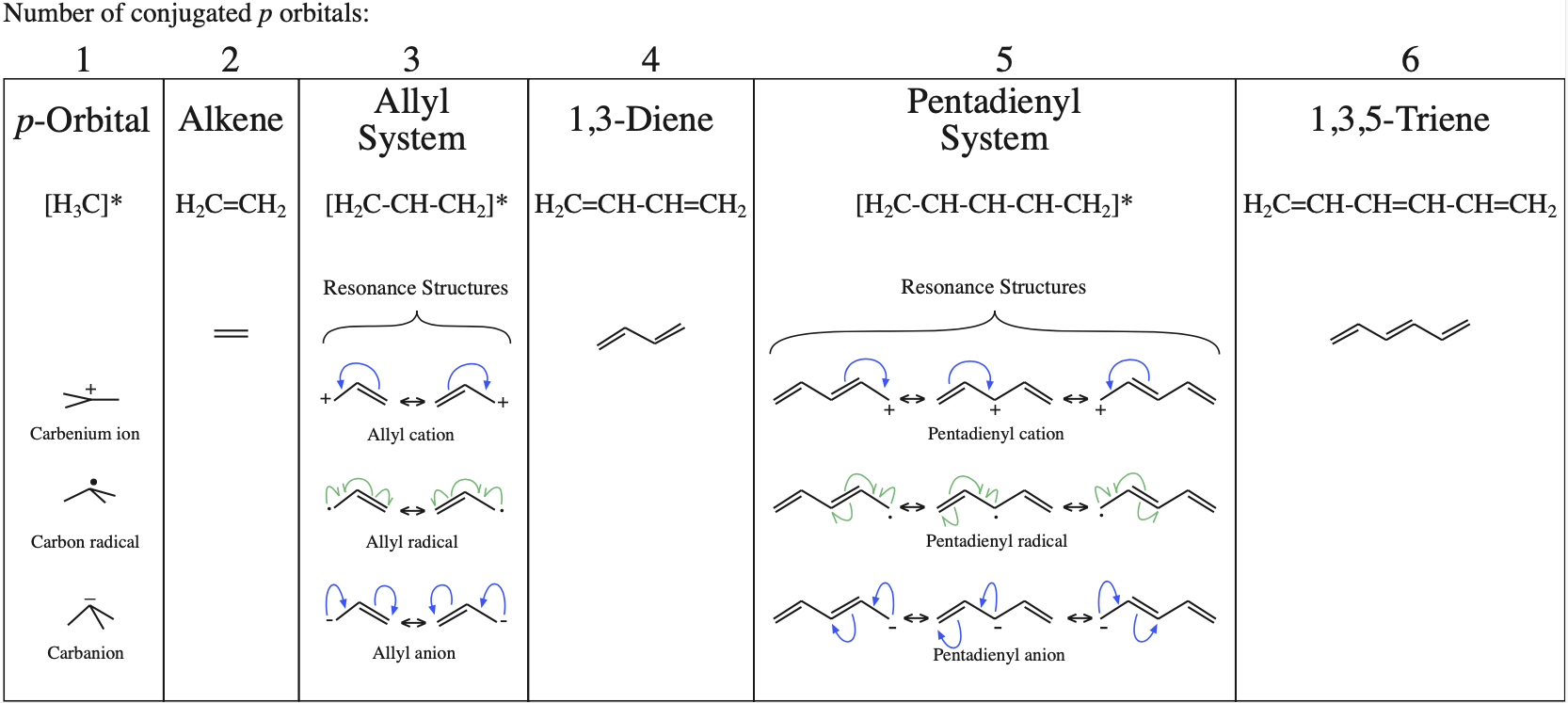
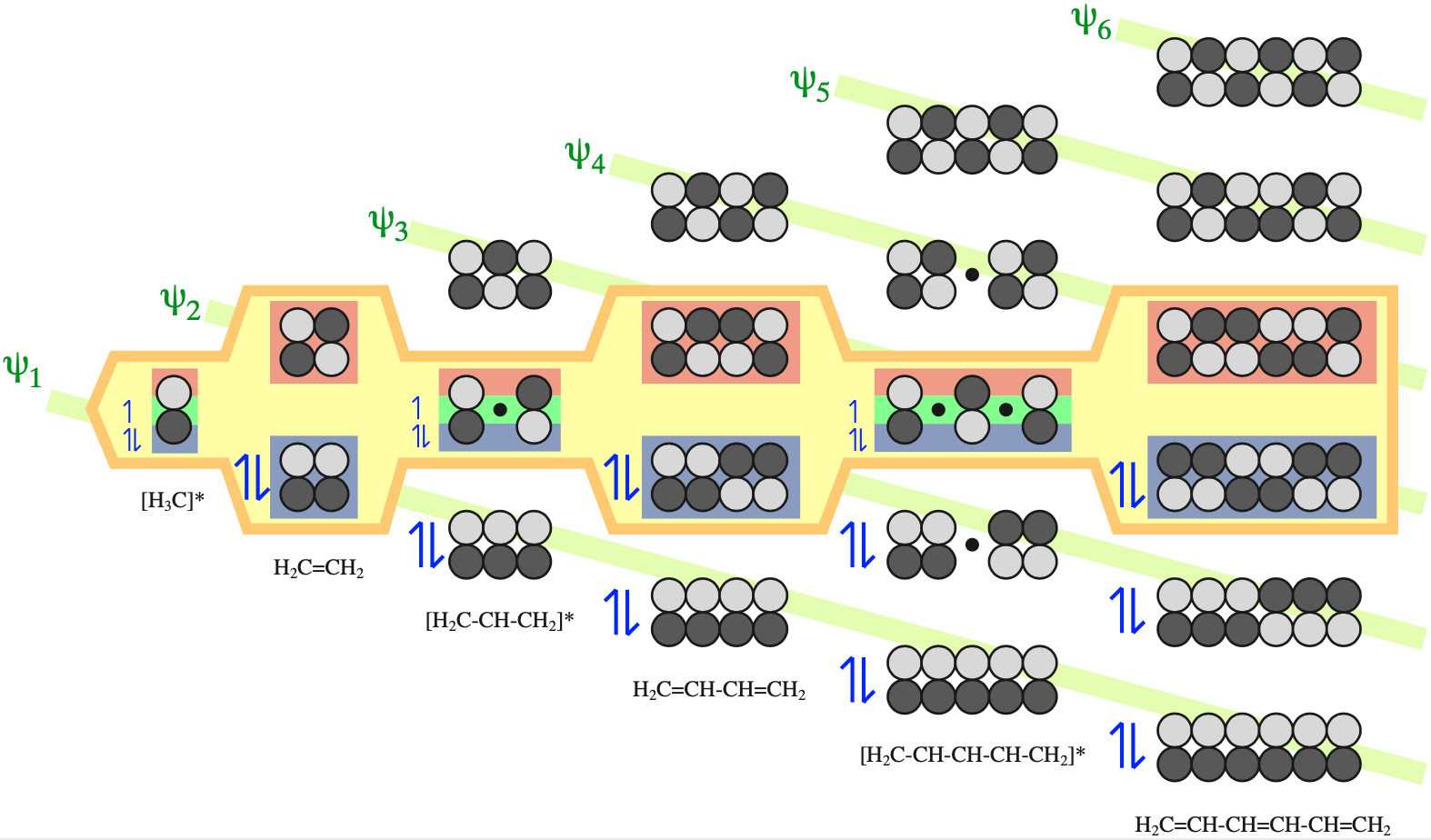
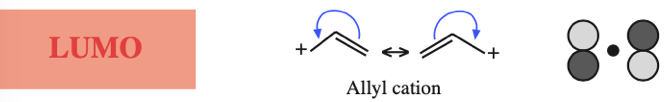
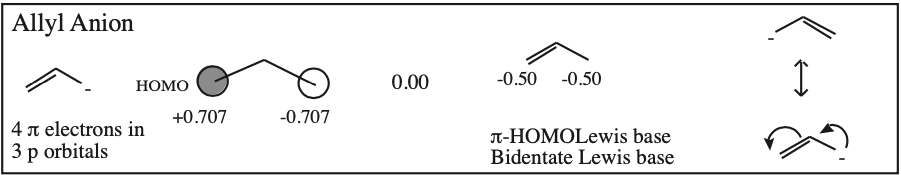
The simplest π-systems are the electronically neutral linear polyene ribbons: ethylene, 1,3-butadiene, 1,3,5-hexatriene, etc. However, the full series includes the isolated p-orbital, the allyl system of three adjacent p-orbitals and the pentadienyl system of five adjacent p-orbitals.
Some points:
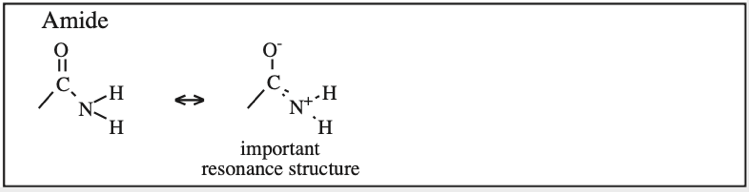
Electrons in π-FMOs are delocalised over all adjacent p–orbitals.
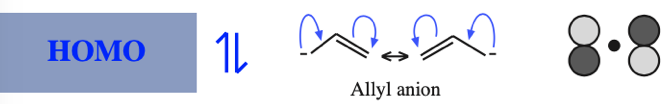
In the VB model, localised alkene type double bonds, anion, cation and radical centres are deemed to exist.
For a given π-MO structure there may be many possible VB resonance structures.
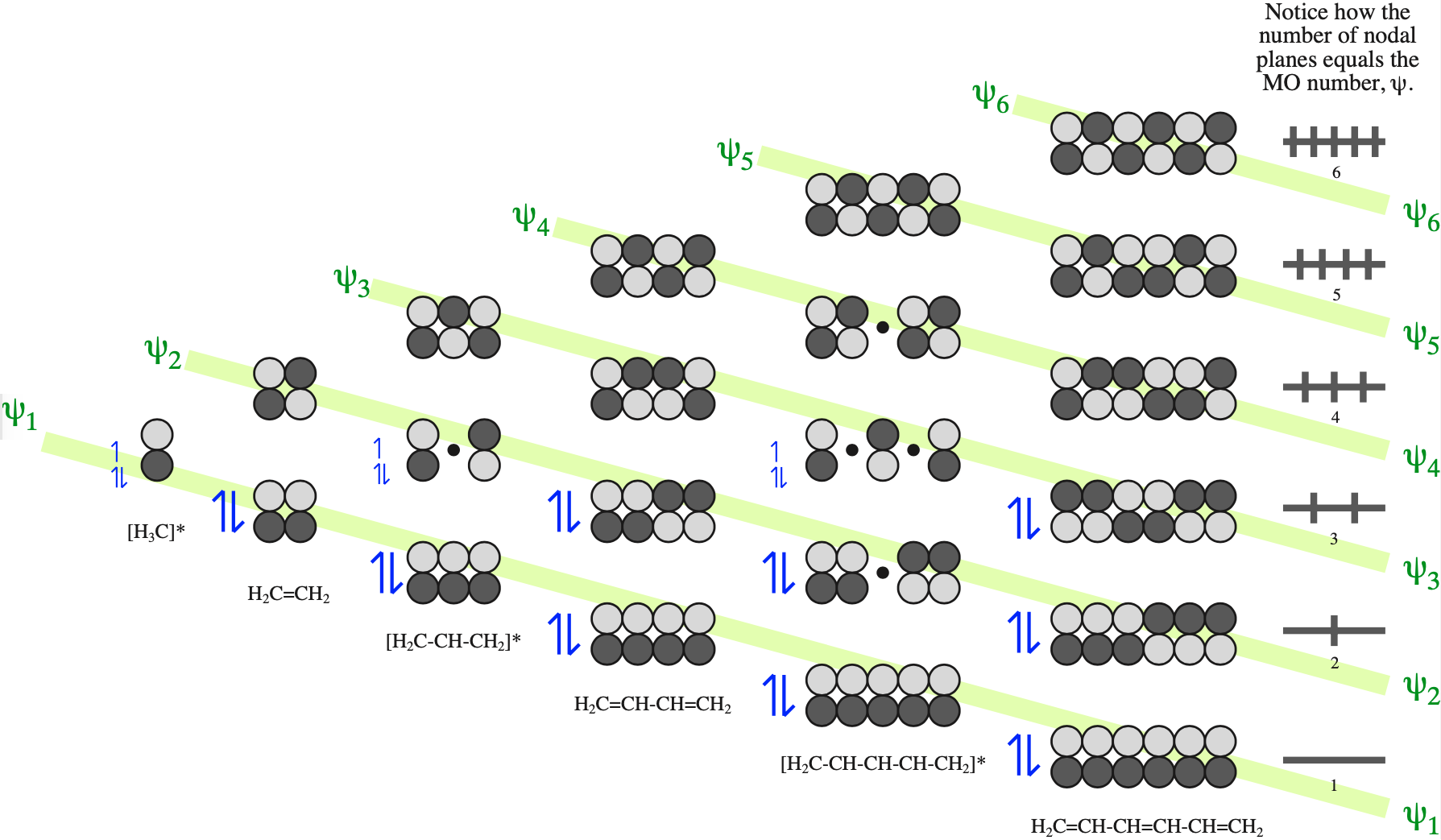
The π MOs form regular patterns. Polyene ribbon π-systems are arranged so that the lowest energy ψ1 MOs have p orbital phases which match right along the "top" and the "bottom" of the system. As the polyenes grow in length, more electrons are required to retain near (+1/–1) electrical neutrality. Electrons enter the increasing energy ψ2, ψ3... MOs in pairs according to the Aufbau principle and Hund's rule.
As energy increases, each MO has one more node (along the length of the polyene ribbon) than the previous MO. At a node there is a phase change.
With alkenes, dienes and trienes all the nodes always occur between atoms, but with the three p orbital allyl system and the five p orbital pentadienyl systems, the nodes sometimes occur at an atom. As a node corresponds to a region of zero electron density these MOs are designated as non-bonding orbitals. Two points:
- It is common to show the positions of these nodal atoms with a "dot".
- Bonding MOs always have corresponding antibonding MOs, but non-bonding MOs do not.
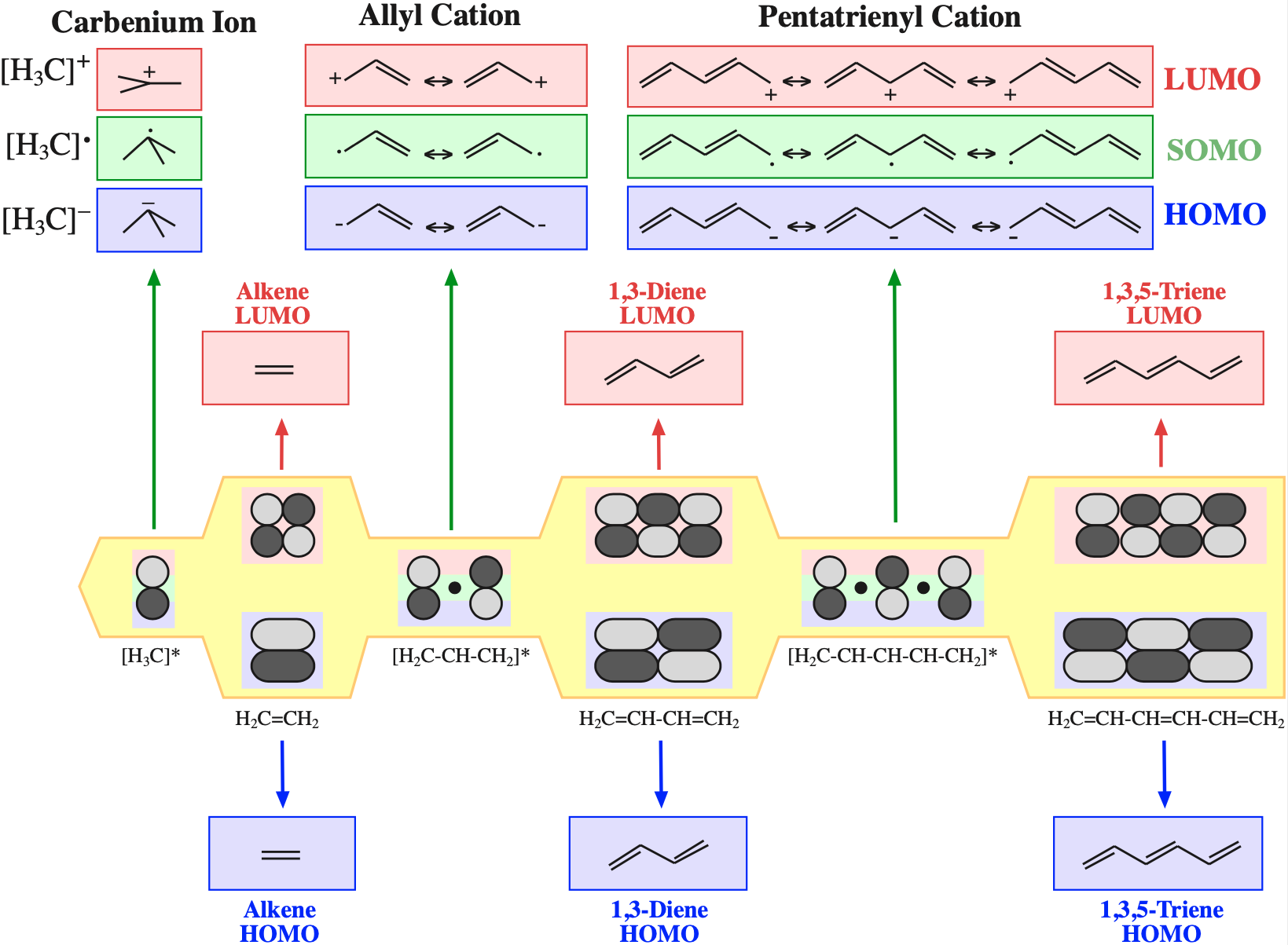
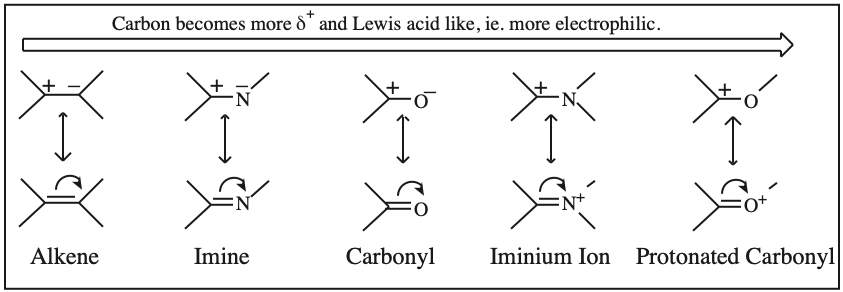
Frontier molecular orbital theory stresses the importance of an FG's HOMO, LUMO and SOMO.
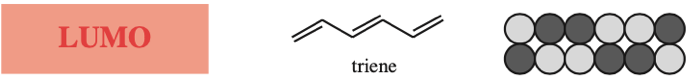
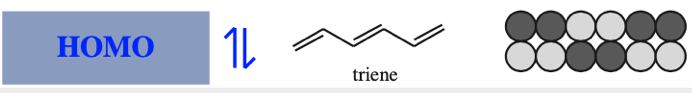
- Alkenes, dienes and trienes are electronically neutral species which contain two, four and six π-electrons respectively.
- The HOMO and LUMO phase characteristics of these polyene ribbon systems can be easily determined from the diagrams.
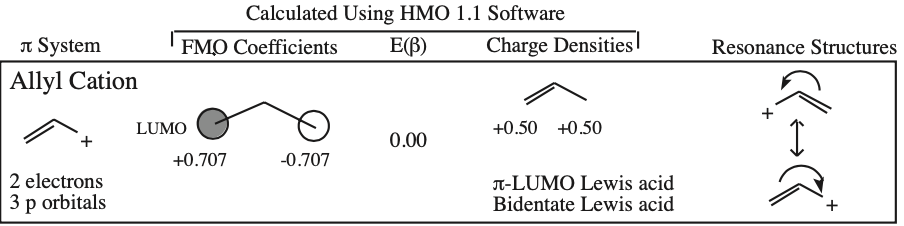
- Allyl cations, radicals and anions have their respective LUMOs, SOMOs and HOMOs associated with psi2.
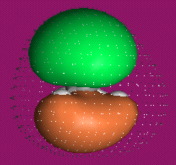
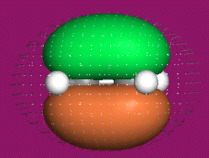
One p-Orbital, Alkene, Allyl System, 1,3-Diene, Pentadienyl System, 1,3,5-Triene:
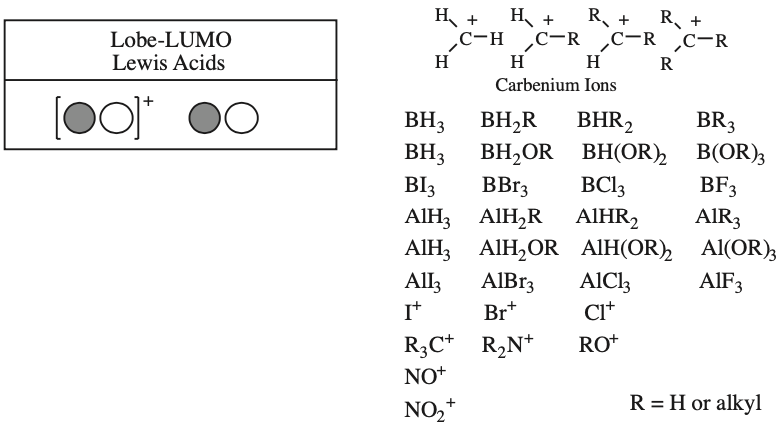
One p-Orbital Functional Groups

Calculated at the Hartree-Fock 6-31G* level using Spartan.



Two Conjugated p-Orbital Functional Groups
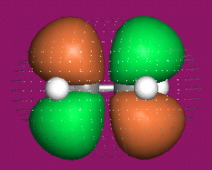


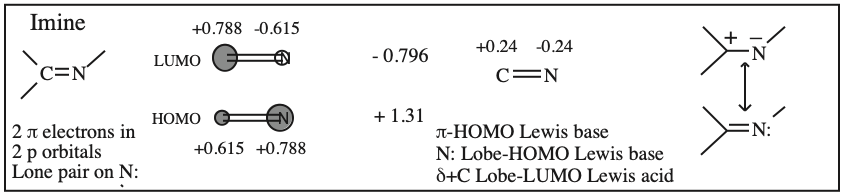
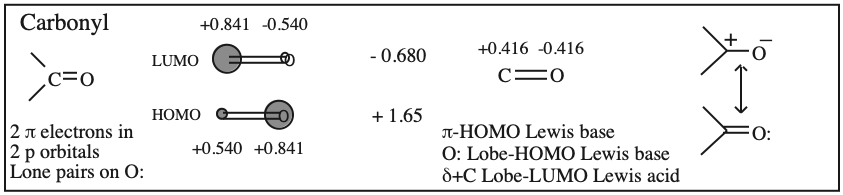
Iminium Ion & Protonated Carbonyl:
Three Conjugated p-Orbital Functional Groups
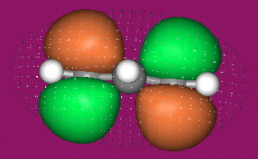

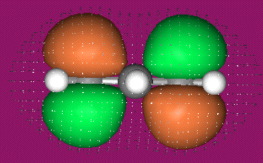
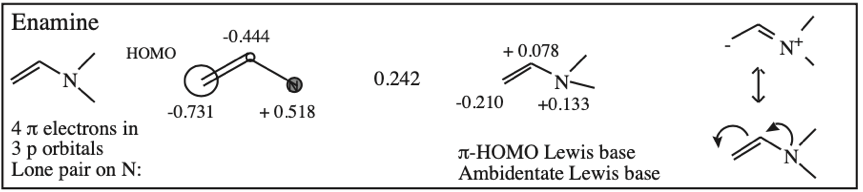
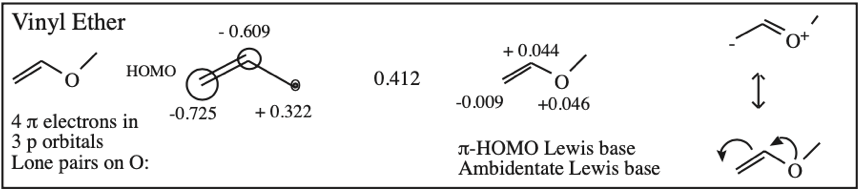
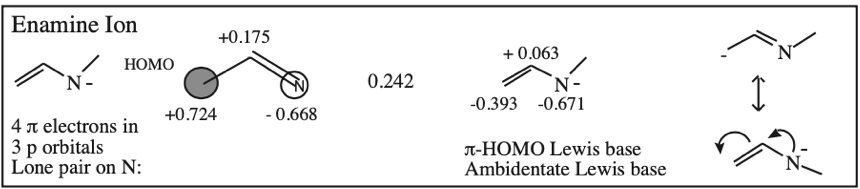

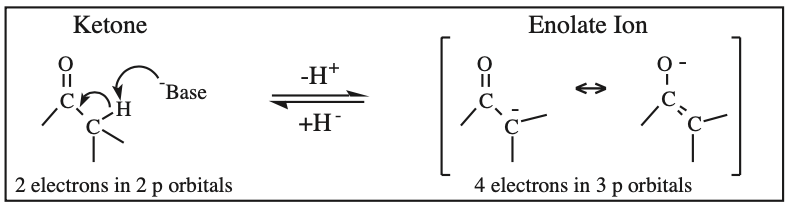
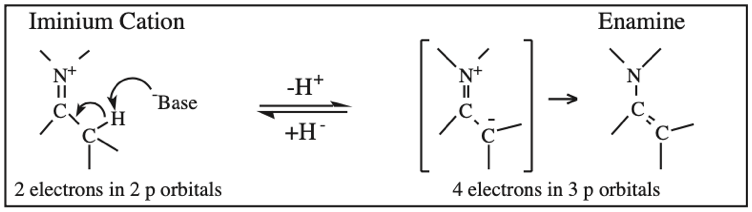
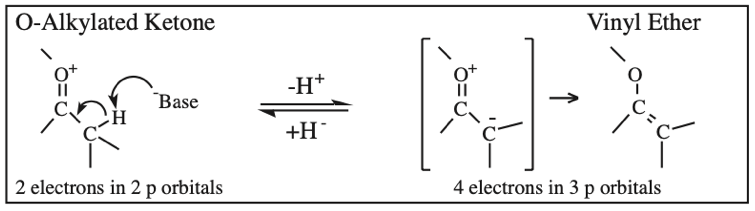
O-Alkylated Ketone / Vinyl Ether:
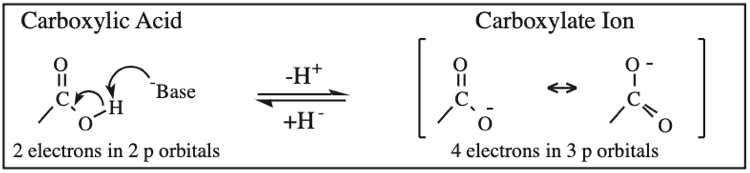
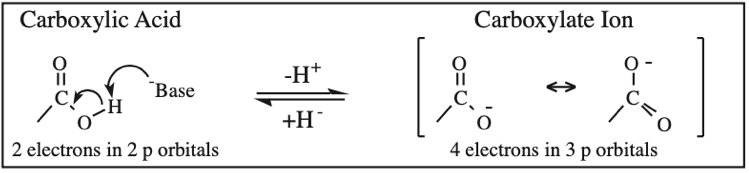
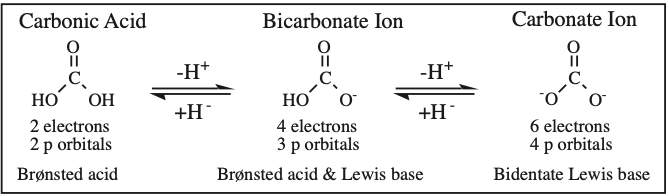
Carboxylic Acid, Carboxylate Anion, Carboxylate Dianion:
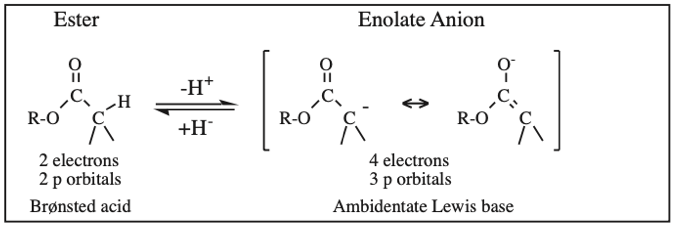
Two + Two Conjugated p-Orbital Functional Groups
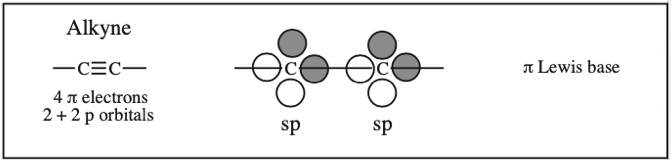

Protonated HCN, Hydrogen Cyanide, Cyanide Ion:
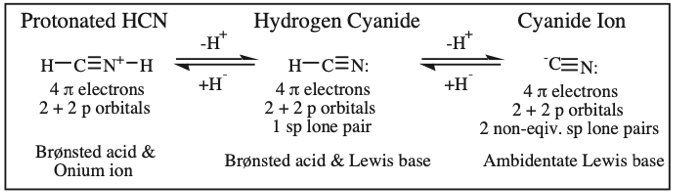
Protonated Alkyl Nitrile, Alkyl Nitrile, Nitrile α-Carbanion:
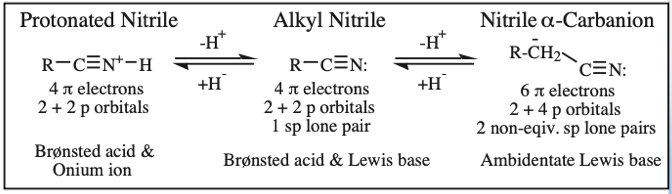
Protonated Nitrogen, Nitrogen (Dinitrogen)
Two + Two Conjugated p-Orbital Functional Groups: Cumulenes
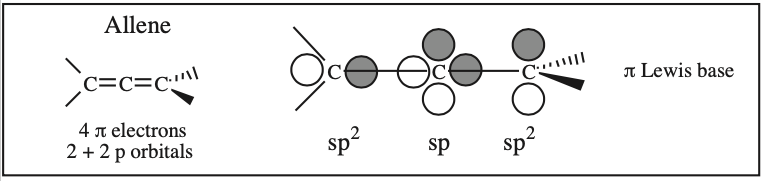
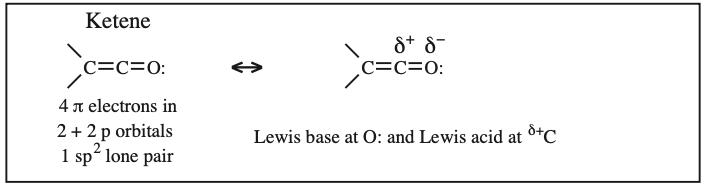



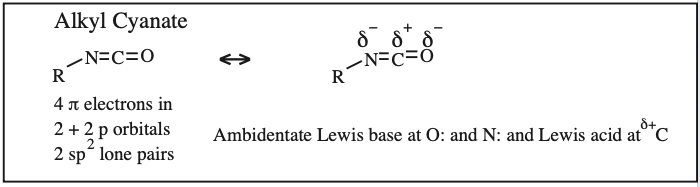
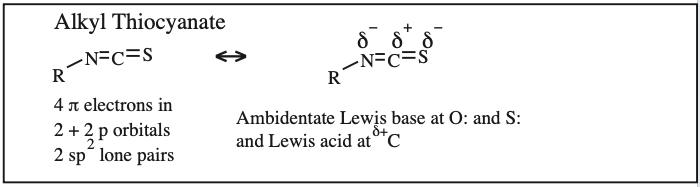
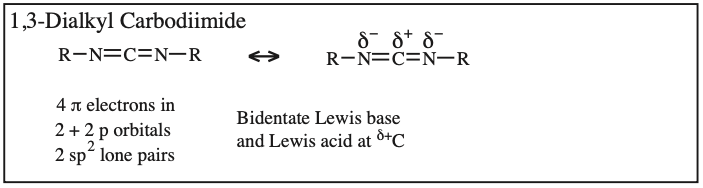
Alkyl Isocyanate, Alkyl Isothiocyanate, Alkyl Azide, Hydrazoic acid:
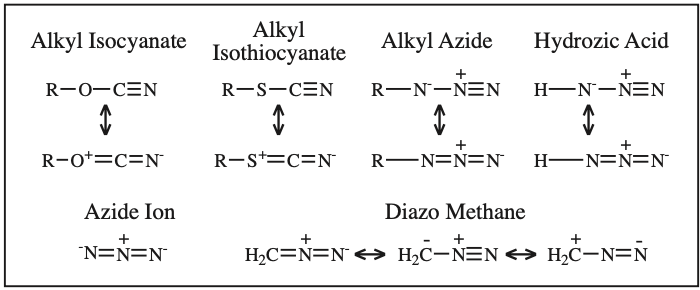
Four Conjugated p-Orbital Functional Groups
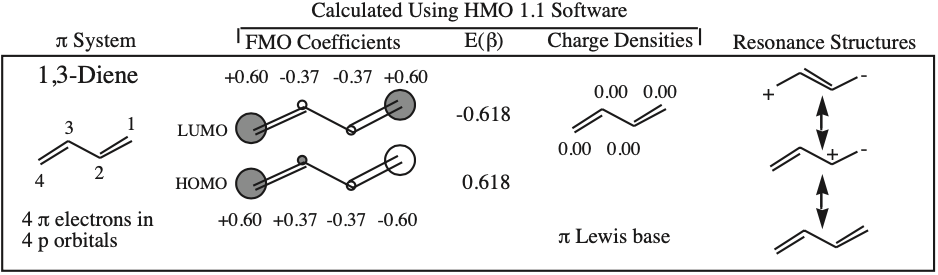
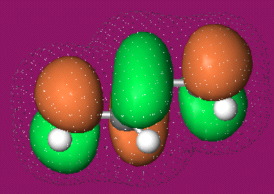
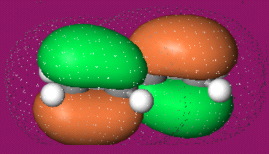
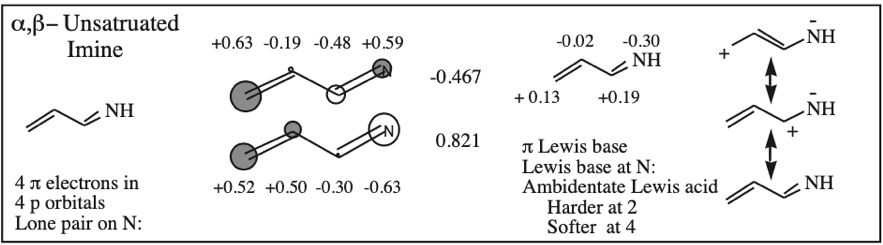
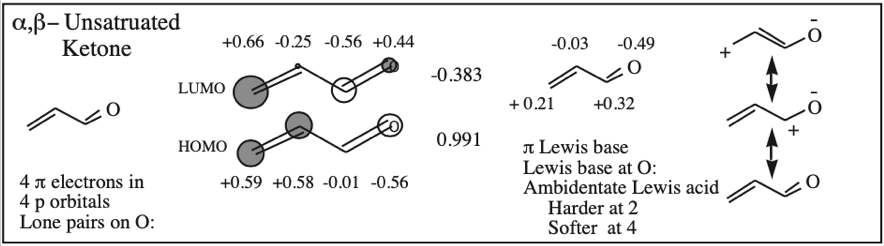
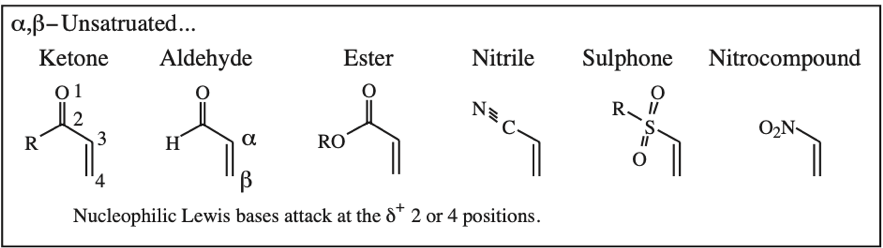
α,β-Unsaturated Ketone, Aldehyde, Ester, Nitrile, Sulfone & Nitrocompound:
Five Conjugated p-Orbital Functional Groups
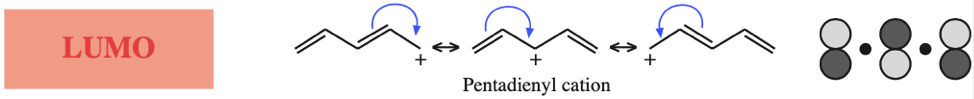
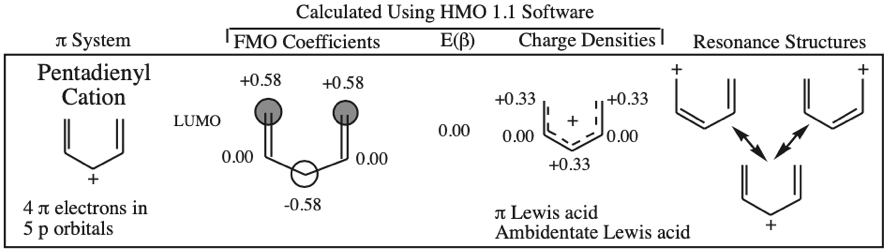
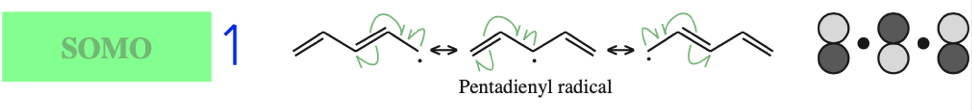
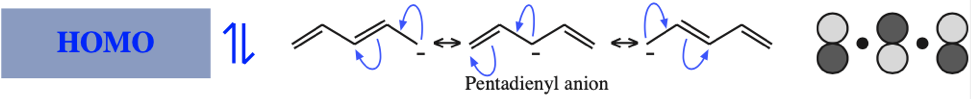
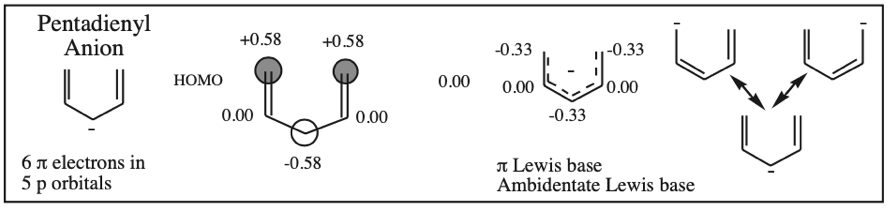
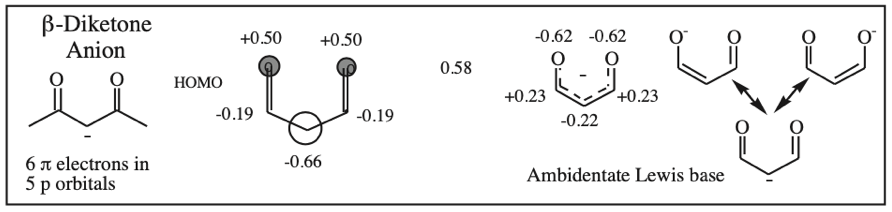
β-Diketone, β-Ketoester, Dialkyl Malonate, β-Ketoacid, etc:
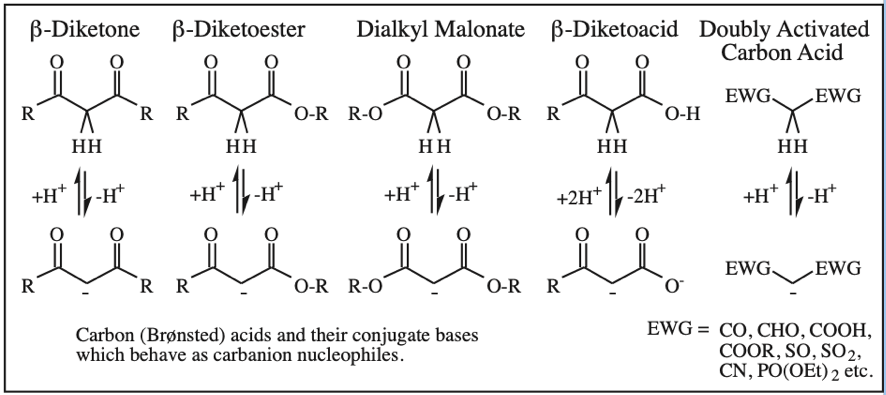
Six Conjugated p-Orbital Functional Groups
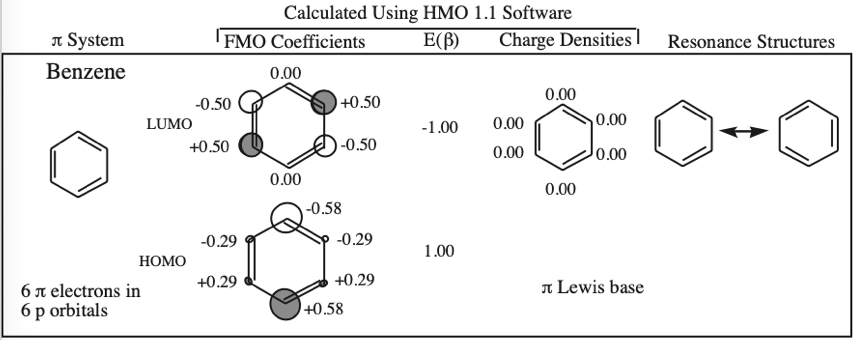
Cyclic Array of p-Orbitals Containing 4n+2 Electrons: Aromaticity
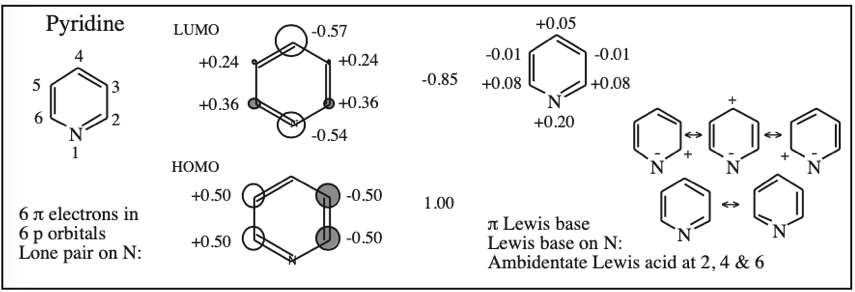
Some unsaturated organic ring systems, such as benzene, C6H6, are unexpectedly stable and are said to be aromatic. A quantum mechanical basis for aromaticity, the Hückel method, was first worked out by physical chemist Erich Hückel in 1931.
In 1951 von Doering succinctly reduced the Hückel analysis to the "4n + 2 rule".
- Aromaticity is associated a cyclic array of adjacent p-orbitals containing 4n+2 π-electrons, where n is zero or any positive integer.
- Aromaticity is associated with cationic, anionic and heterocyclic π-systems, as well as neutral hydrocarbon structures like benzene and naphthalene.
- Aromaticity can be identified by a ring current and associated down-field chemical shift in the proton NMR spectrum. (Aromatic compounds have a chemical shift of 7-8 in the proton spectrum).
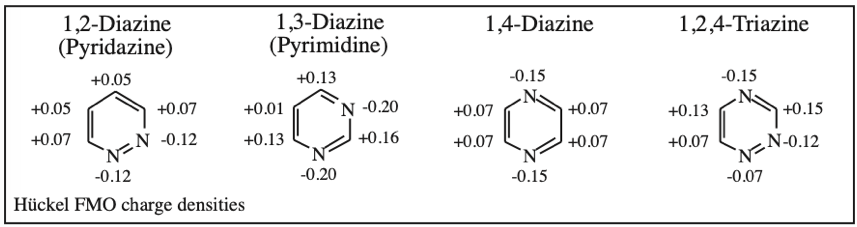
1,2-Diazine (Pyridazine), 1,3-Diazine (Pyrimidine), 1,4-Diazine (Pyrazine) & 1,2,4-Traizine:
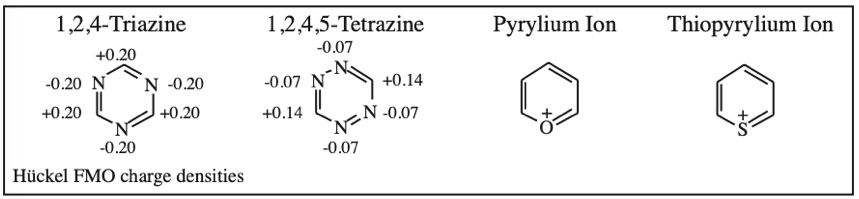
1,3,5-Traizine, 1,2,4,5-Tetrazine, Pyrylium Ion & Thiopyrylium Ion:
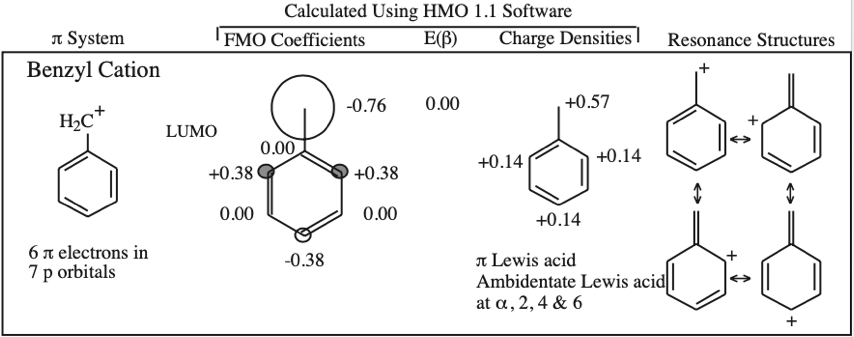
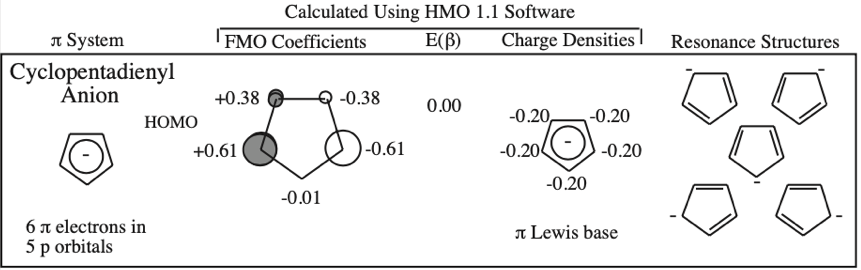
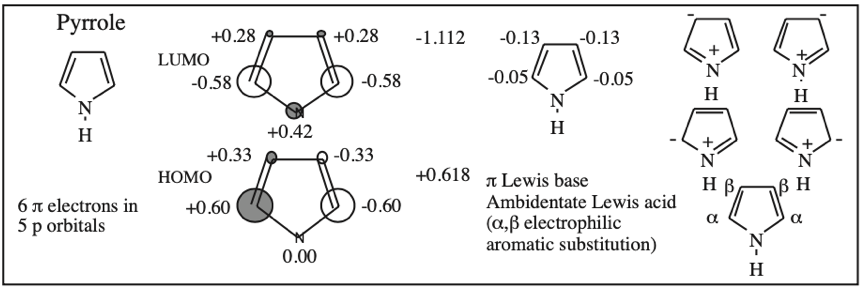
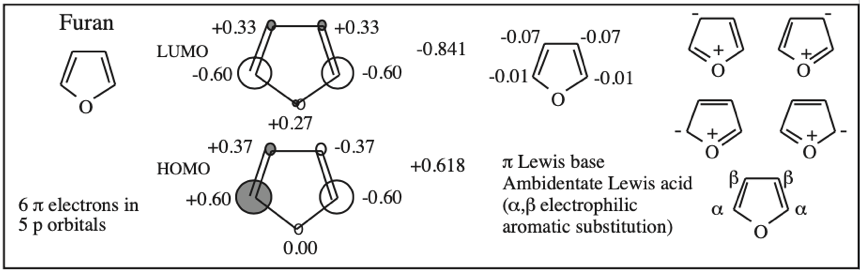
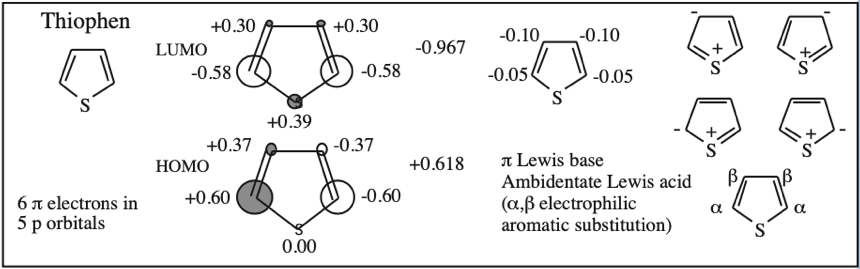
Some Exotic Aromatic Systems
- –H+ means remove a proton to form the conjugate base.
- –Nfg– means remove a nucleofugal Lewis base like Cl–, TsO–, etc. to form the 'conjugate cation'.
Cyclopropenyl Cation:

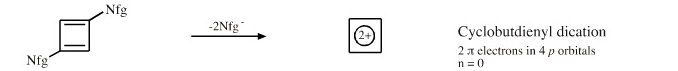
Cyclobutadienyl Dication:



Cyclooctatetraenide Cation:




Methylene Bridged Cyclodecapentaene:


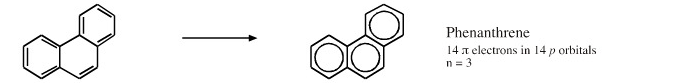
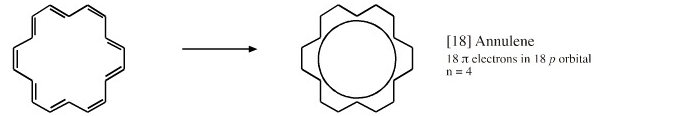
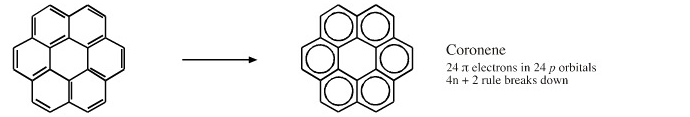
Substituted Benzenes and Susceptibility to Electrophilic Aromatic Substitution:
- Electron donating functions, such as the -OH group, activate a benzene ring towards SEAr reactions and are ortho, para (2, 4, 6) directing.
- Electron withdrawing groups, such as the -NO2 nitro group, deactivate the benzene ring towards SEAr and are meta (3, 5) directing, but they activate towards SNAr assuming there is a suitable leaving group present, such as a halogen.
Activating and directing effects can be parameterised using carbon-13 NMR data, specifically the 13C chemical shift of the para-carbon (4-position) of a mono-substituted benzene. The effect is more easily visualised if the chemical shift data is quoted with respect to benzene rather than TMS.
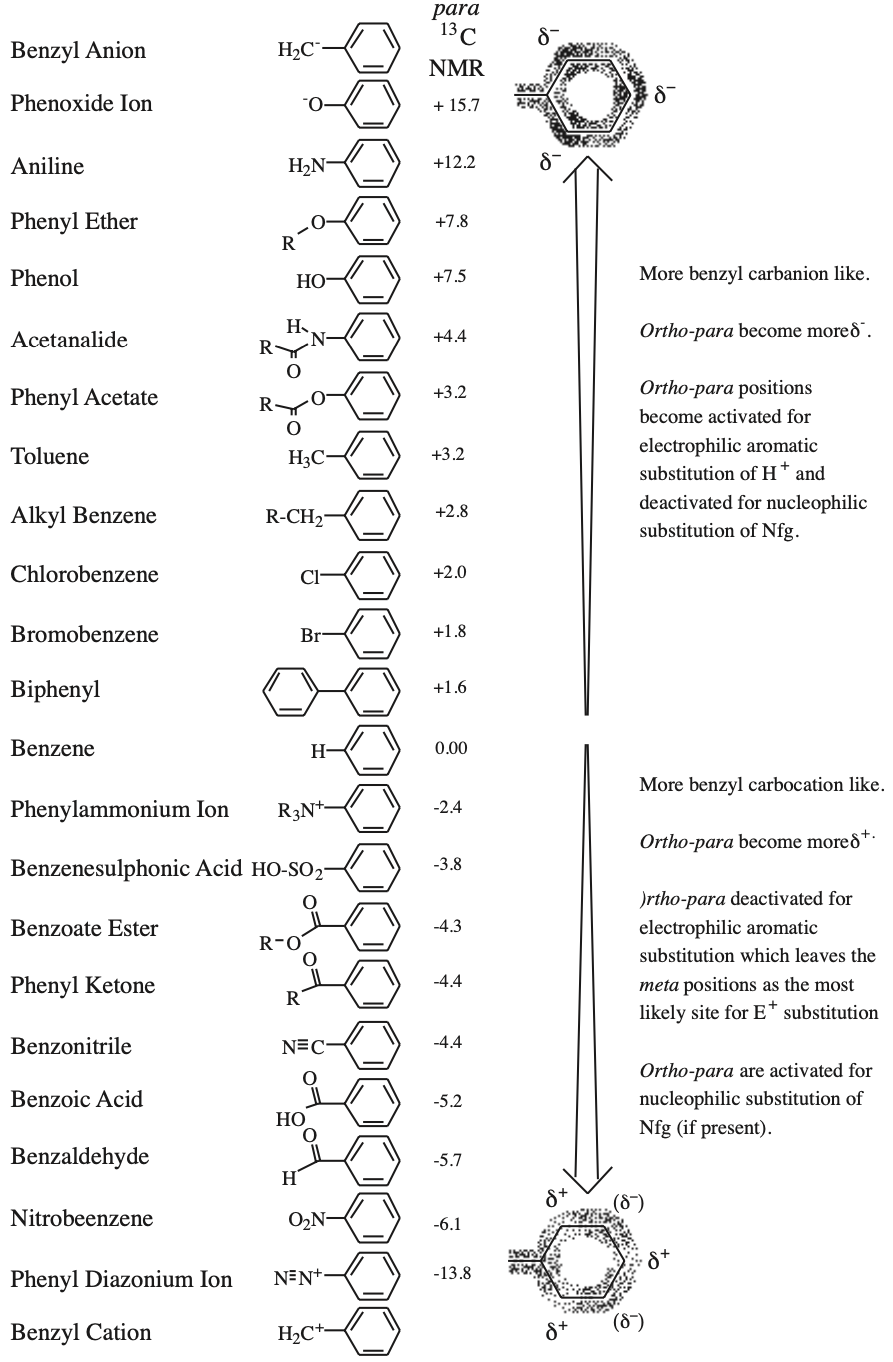
Species with para-carbons “up-field” of benzene (negative chemical shift values) are electron rich at the para 4-position and are assumed to be electron rich at the ortho (2 & 6) positions as well. The 2, 4 & 6 positions are correspondingly susceptible to electrophilic aromatic substitution. The more “up-field” the para-carbon is, the more activated the compound is for ortho-para directed SEAr.
Species with para-carbons “down-field” of benzene (positive chemical shift) are electron poor at the ortho-para (2, 4 & 6-) positions making the meta (3 & 5-) positions relatively electron rich. The effect is that the meta positions are the more susceptible to electrophilic substitution, although these positions are deactivated w.r.t. benzene.
If a system with a down-field para-carbon has a nucleofugal leaving group (usually chloride or bromide) ortho or para to the main function, the Nfg functions are susceptible to nucleophilic aromatic substitution, SNAr. Again the greater the down-field shift w.r.t. benzene the more pronounced the effect, assuming that no other reaction pathway predominates.
These effects are additive: two or three electron donating or electron withdrawing groups will have more influence than one.
Why The para-Carbon?
13C NMR spectroscopy, briefly, measures the electronic environment of carbon atoms. For aromatic carbon atoms there are at least three electronic environmental components which contribute to the chemical shift of a particular carbon atom:
- The aromatic ring-current which has the effect of sending aromatic carbons down-field by 100ppm or so.
- Perturbations to the aromatic ring current caused by attached functional groups acting through the π-system.
- Direct, local and “through-space” effects caused by adjacent functional groups. Unfortunately, these cause very large perturbations to the chemical shift of ortho (and to a lesser extent meta) carbons.
|
|
| Polyatomic Species: Hybrid & Molecular Orbitals | Functional Group Database |
© Mark R. Leach 1999 –
Queries, Suggestions, Bugs, Errors, Typos...
If you have any:
Queries
Comments
Suggestions
Suggestions for links
Bug, typo or grammatical error reports about this page,please contact Mark R. Leach, the author, using mark@meta-synthesis.com
This free, open access web book is an ongoing project and your input is appreciated.