Periodic Table |
 |
 |
 |
 |
 |
 |
| Clickable Lewis Acid/Base Matrix | Redox Chemistry |
Nucleophiles | Bases | The Organic Chemistry of The Fluoride Ion
Congeneric array interaction logic can be used to explore the nucleophilic substitution behaviour of the organohalides, R–F, R–Cl, R–Br and R–I. Using bond length as a proxy for hardness, the behaviour of the anomalous fluoride ion can be explained. This page is concerned with Type 15 Lewis acid/base complexation chemistry:
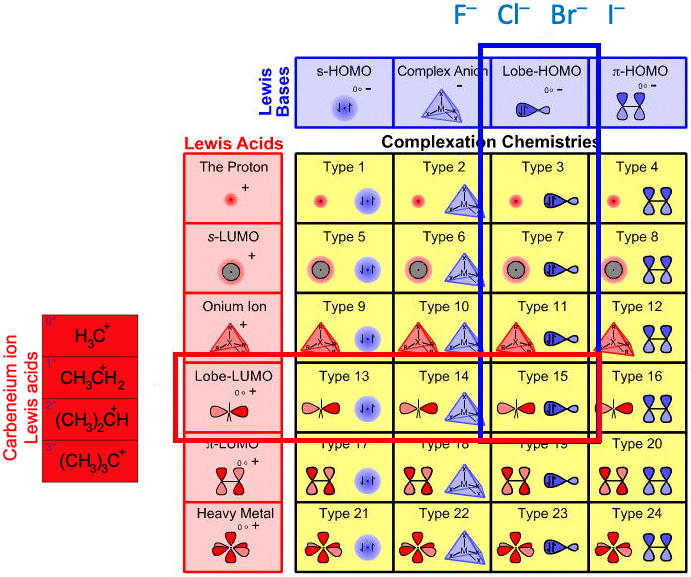
Alkylhalides
The chloride, bromide and iodide ions are important organic chemistry moieties, and they can be added and removed using the various addition, elimination and substitution reactions available to organic chemists.
Generally, in organic chemistry Cl–, Br– & I– behave as a congeneric series of Lobe-HOMO Lewis bases: chloride ion is quite a good nucleofuge or Lewis base leaving group, bromide a better leaving group (the C-Br bond is more labile) and the iodide ion better still:
Cl– > Br– > I– or Cl– < Br– < I–
But fluoride ion does not fit into this series, and F– is seldom displaced from alkyl carbon centres by a nucleophile.
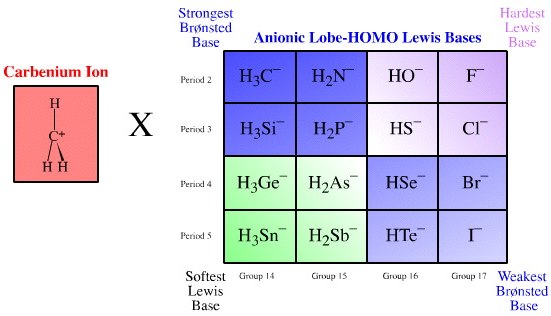
We can explore this apparent fluoride ion anomaly by examining the interaction of the Lobe-LUMO carbenium ion, H3C+, Lewis acid with the anionic Lobe-HOMO Lewis base congeneric planar (as discussed here):
This (series x dot) congeneric interaction is rather like the proton, H+, interaction with the same anionic Lewis bases, here. That planar was congeneric with respect to both pKa and bond length over the entire planar, even though pKa and "to-–hydrogen" bond length data pointed in different directions.
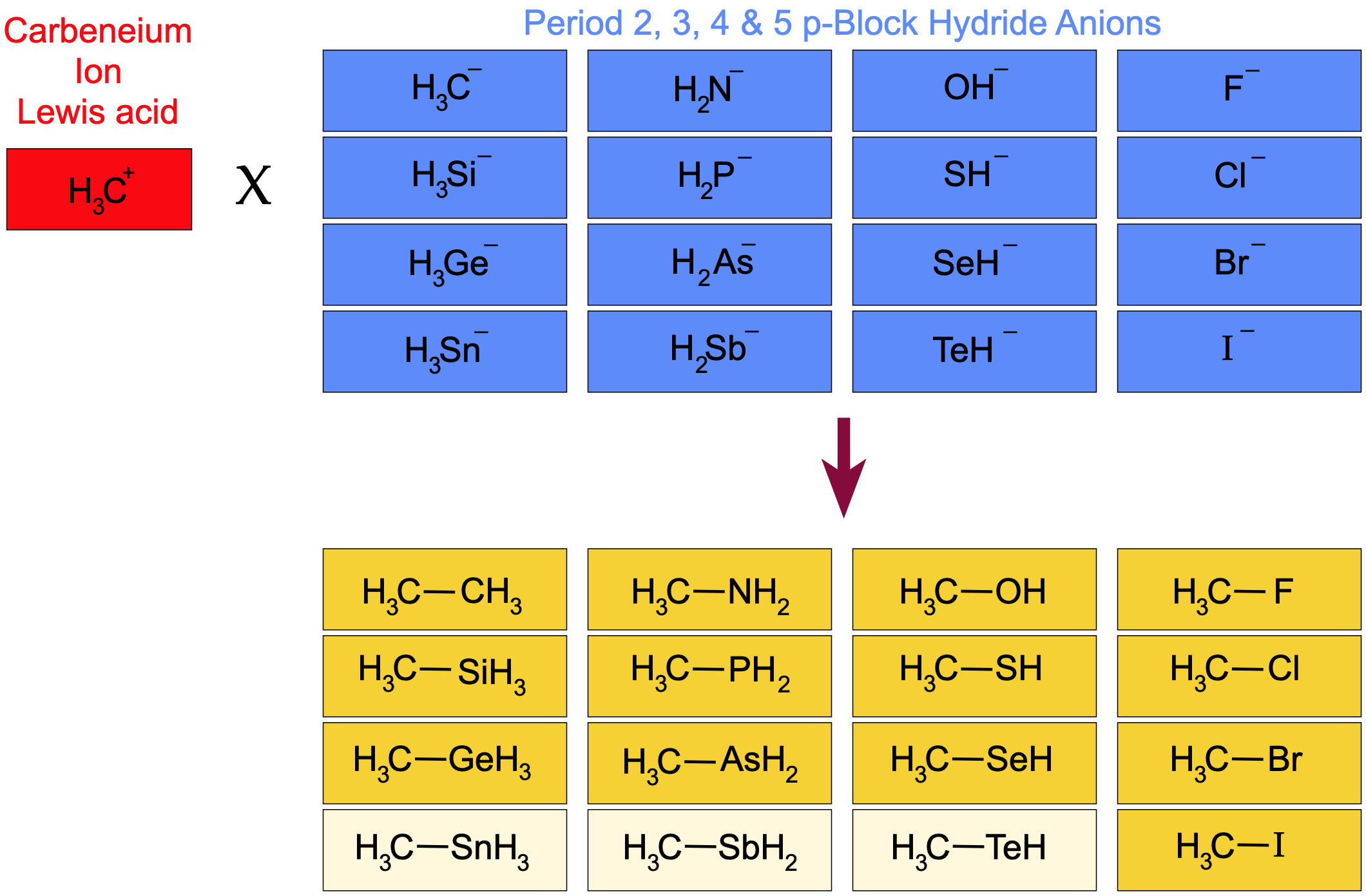
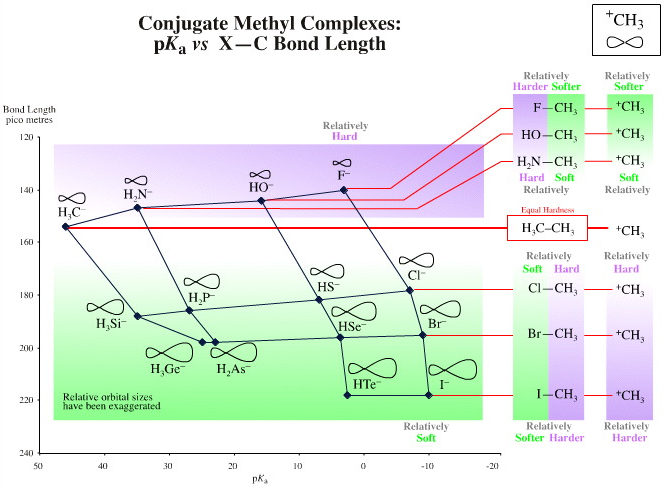
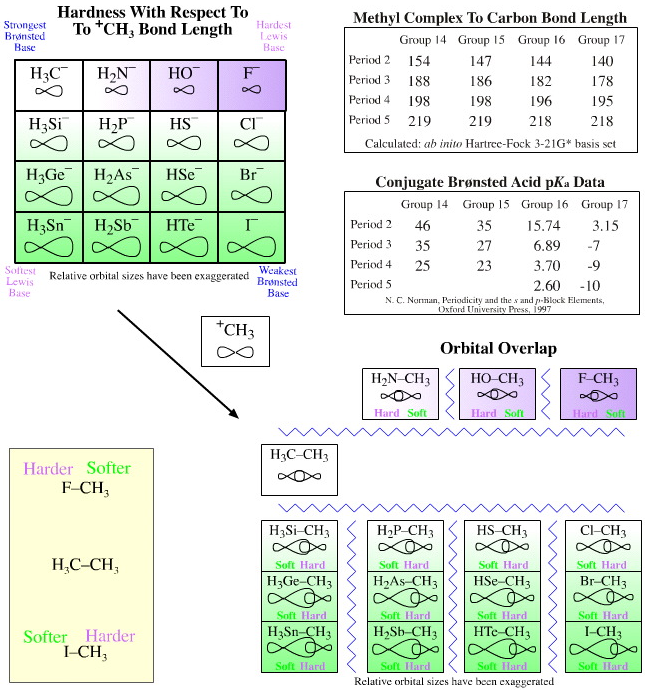
When we plot the "to–CH3" bond length data instead of "to–H" bond length, again against pKa, the situation is a little different due of the nature of the C–C bond (formed by H3C+ plus –CH3) and its central importance to organic chemistry. We find that if we use the C–C bond length (154pm) as a standard:
- The C–F bond length (140pm) is shorter
- The C–Cl, C–Br and C–I bond lengths (178, 195, 218pm) are longer
Let us resurrect the Pearson notion of hardness and ask the question: "Which end of the H3C–, H2N–, HO–, F– congeneric series is harder?"
- The methyl anion H3C– (as methyl lithium, H3CLi) is a super-base. On the Ka scale the methyl anion proton abstracts hydrogen some 43 orders of magnitude more aggressively than the fluoride ion F– does. (Conjugate acid data: CH4Ka = 1 x 10–46, HF Ka = 1.8 x 10–4).
- However, the fluoride ion has a shorter "to–H" bond length with its conjugate Brønsted acid than does the methyl anion (HF = 94 pm, CH4 = 108 pm).
For this system, hardness is here (on this web age) defined with respect to bond length.
- Short "to H+" bond length equates with hard
- Short "to H3C+" bond length equates with hard
- Long "to H+" bond length equates with soft
- Long "to H3C+" bond length equates with soft
Due to the special nature of the C–C bond and its importance to organic chemistry, the methyl cation H3C+ is of the same hardness as the methyl anion H3C–.
- The fluoride ion F– has a shorter "to H3C+"
- bond length than H3C– so F– is harder than H3C–. F—CH3 = 140 pm, H3C—CH3 = 154 pm
- However, the chloride, bromide and iodide anions have longer "to H3C+" bond lengths than H3C–, so the heavier halogen anions are softer Lewis bases than H3C–.
Cl—CH3 = 178 pm, Br—CH3 = 195 pm & I—CH3 = 218 pm
These structural trends are mirrored in the SN1, SN2, El and E2 reaction chemistry at carbon centres. Fluoride ion’s, F–, behaviour is linear with respect to its congeneric siblings HO–, H2N– and H3C–, but not with respect to Cl–, Br– and I–.
- The fluoride ion F– is moderately nucleophilic (F– < HO– < H2N– < H3C–) compared with oxyanions, nitranions and carbanions (below), yet it seldom behaves as a nucleofugal Lewis base leaving group, although it is the best compared with its congeneric siblings (F– > HO– > H2N– > H3C–).
- On the other hand, chloride, bromide and iodide are generally rather poor nucleophiles at carbon centres, but they make excellent nucleofuges (Cl–< Br–< I–).
Thus, organic chemistry with its special emphasis on the C–C bond has the fluoride ion, F–, harder than the carbanion at delta+ carbon centres whereas the chloride, bromide and iodide are softer.
Carbanions, Nitranions, Oxyanions and Thioanions
In the section above we used "to CH3" bond length, but the same logic can be used with "to H" bond length because the data sets have such similar form:
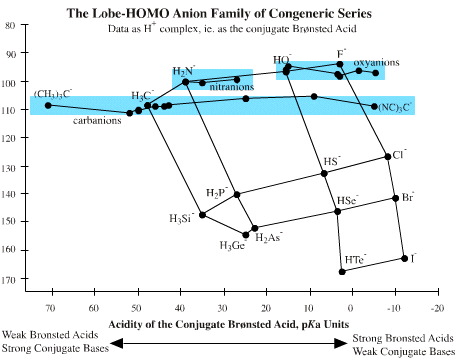
The "to H" bond length of the conjugate Brønsted acid correlates with intrinsic nucleophilicity, a measure of reactivity towards delta+ carbon centres. In organic chemistry the C–H bond is ~108 pm.
- Fluoride ion, oxyanions and
nitranions are harder than carbanion
- Thioanions, chloride ion, bromide ion and iodide, etc. are softer than carbanion
A Brønsted acid's pKa is a measure of the conjugate base's ability to abstract protons. Due to the rapid nature of proton transfer, if a base is able to abstract a proton, it will. Or to put this another way, the presence of a Brønsted acid will quench a basic centre, both as a Brønsted proton abstracting base and as a nucleophilic Lewis base.
- For example, many of the anions we are discussing in this section are far more basic than water, and water will quickly destroy any strongly basic anions present. A methyl anion will rapidly react with moisture to give methane plus hydroxide ion (or even oxide ion). These reactions have to be carried out in non-aqueous solvents, usually diethyl ether or THF.
The pKa of oxyanions and nitranions and carbanions, particularly carbanions, can vary widely, as the graph above shows.
An anionic Lewis base, X–, has several reactivity choices, it can act as a:
- Nucleophile, Nu–,
and displace (substitute) a nucleofugal Lewis base leaving group, Nfg–
- Brønsted base, B–,
and abstract an available proton
- Spectator counter ion and do nothing at all
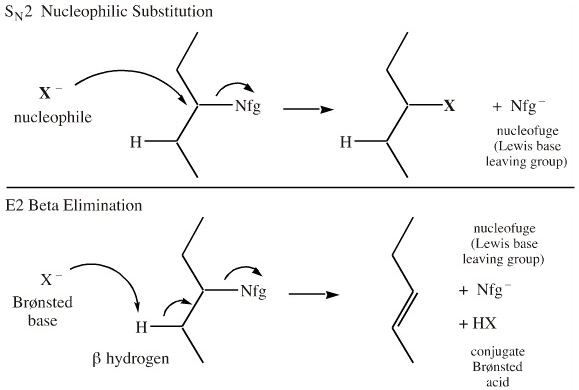
These reaction pathways are highly pertinent in organic chemistry.
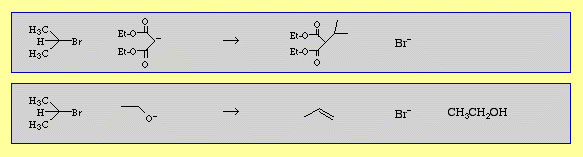
2-bromopropane (isopropyl bromide) undergoes nucleophilic substitution with the diethylmalonate carbanion but beta-elimination with hard ethoxide ion.
It is possible to have carbanion and oxyanion centres in the same function, as seen with the enolate ion:
The hard oxygen end of an enolate ion will preferentially react with triethyl oxonium ion (a donor of hard Et+) whereas the carbanion end will react with ethyliodide (a donor of softer Et+).
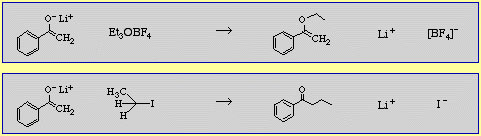
A selection of examples of this type are contained in The Chemical Thesaurus reaction chemistry database.
Carbanions
There is the greatest variety of behaviour amongst the carbanions, which range from super proton abstracting bases (such as the alkyl lithium reagents) to highly efficient nucleophiles.
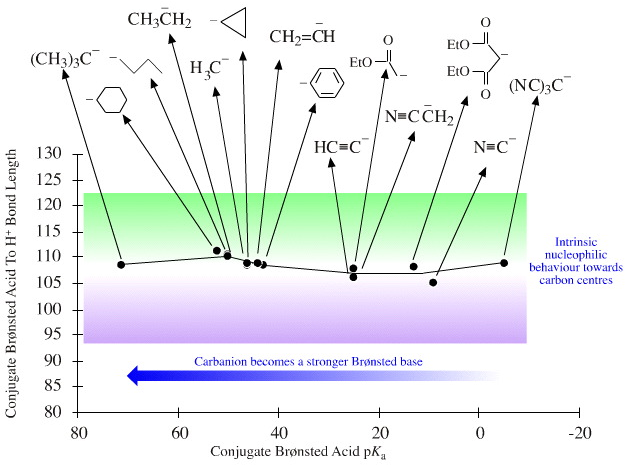
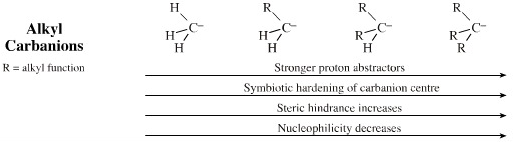
Using the methyl anion, H3C–, as the reference species:
Adding alkyl functions destabilises the carbanion centre and increases pKa (proton abstracting ability). However, the alkyl groups also cause steric hindrance around the carbanion centre which decreases the ability to reach a delta+ carbon. As a result adding alkyl functions decreases nucleophilicity. Tse-Lok Ho suggested that tertiary alkyl carbanions are harder than the methyl carbanion.
Adding adjacent benzene ring π-systems stabilises the anion's negative charge with the result that pKa decreases. However, the steric hindrance increases dramatically because aromatic rings are so large and this reduces nucleophilicity.
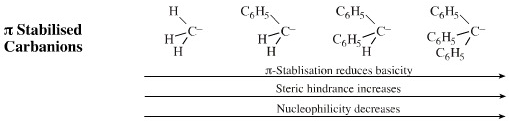
Electron withdrawing functions (ketone, aldehyde, ester, nitrile, nitro) stabilise the adjacent carbanion centre and reduce the pKa of the carbanion centre. Species with one and two electron withdrawing groups, such as enolate ions and diethyl malonate, are amongst the most versatile of all nucleophilic carbanions.
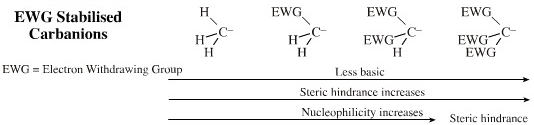
The reactivity of a carbanion can be modified by the nature of the positive counter ion. If we take lithium as a reference point, Na+ and K+ will increase the bond polarisation and make the reagent more aggressive (alkyl sodium and potassium compounds are not stable), but also less soluble (in the case of EWG stabilised carbanions).
- It is usually more useful
to decrease the bond polarisation of alkyl carbanion reagents
with a less electropositive (softer) metal cation ion.
- Magnesium "Grignard reagents" and organozinc compounds are much less [Brønsted] basic than the corresponding lithium reagents and so are able to show their nucleophilic side. (Grignard reagents participate in nucleophilic addition to ketones whereas the more Brønsted basic alkyl lithium reagents will abstract a beta-proton.)
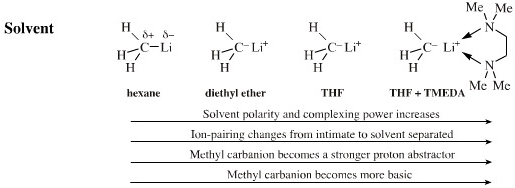
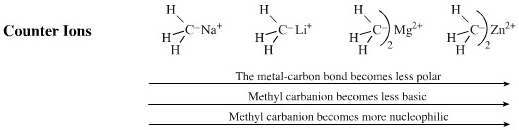 The solvent can influence
the reactivity of a carbanion. A solvent with Lewis base centres is
able to compete to complex lithium (or other) counter ion. Diethyl ether
is better at complexing Li+ than hexane and so the carbanion
is more reactive in diethyl ether than hexane. (There is less ion-pairing and the anion is said to be 'naked'.)
The solvent can influence
the reactivity of a carbanion. A solvent with Lewis base centres is
able to compete to complex lithium (or other) counter ion. Diethyl ether
is better at complexing Li+ than hexane and so the carbanion
is more reactive in diethyl ether than hexane. (There is less ion-pairing and the anion is said to be 'naked'.)
Complexing agents such as tetramethylethylenediamine (TMEDA) or crown ethers can make the carbanion even more reactive. Again, this logic applies to any anionic Lewis base.
Bases in Synthesis
A nice video by YouTuber That Chemist in which he ranks Brønsted bases by their synthetic utility as H+ acceptors. He talks alot about steric hinderance making the Lewis base a better proton acceptor and reducing the base's nucleophilic tendencies, as discussed above:
|
|
| Clickable Lewis Acid/Base Matrix | Redox Chemistry |
© Mark R. Leach 1999 –
Queries, Suggestions, Bugs, Errors, Typos...
If you have any:
Queries
Comments
Suggestions
Suggestions for links
Bug, typo or grammatical error reports about this page,please contact Mark R. Leach, the author, using mark@meta-synthesis.com
This free, open access web book is an ongoing project and your input is appreciated.