Periodic Table |
 |
 |
 |
 |
 |
 |
| Lewis Acids & Lewis Bases | Matrix Poster |
The Lewis Acid/Base Interaction Matrix
School and university students around the world learn that electron pair donor Lewis bases "react with" or "complex with" or "interact with" electron pair acceptor Lewis acids.
- The previous page showed that there are six quite distinct types of Lewis acid and four quite distinct types types of Lewis base, where distinction is by frontier molecular orbital (FMO) topology.
- It follows that the the six types of Lewis acid and the four types of Lewis base inevitably interact to give 24 distinct types of Lewis acid/base complex.
- The chemistry encompassed and described by the Lewis Acid/Base Interaction Matrix is quite staggering. It ranges across organic, inorganic & organometallic reaction chemistry in such a way that each is seen as an inevitable manifestation of the Lewis acid/base interaction.
- The Lewis acid/base interaction matrix is the core finding of the chemogenesis analysis.
Six Types of Lewis Acid and Four Types of Lewis Base
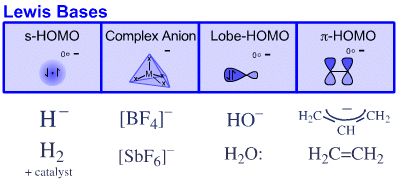
The previous page introduced the notion that there are four distinct types of Lewis base, where classification is based on the frontier molecular orbital FMO topology. These four types of Lewis base are:
s-HOMO Lewis bases: H–, H2
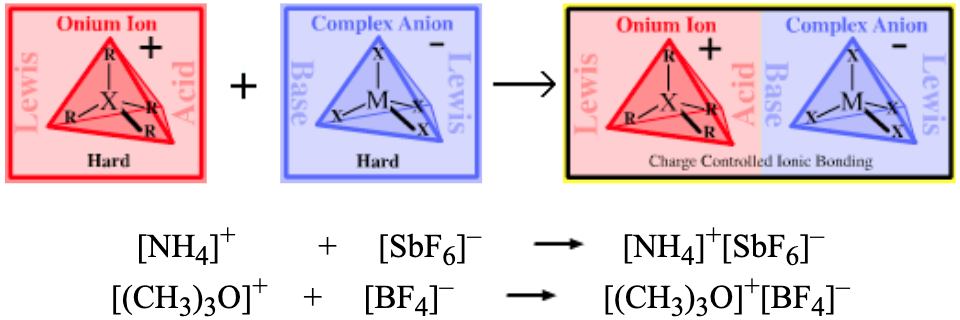
Complex Anion Lewis bases: [BF4]–, [SbF6]–, etc.
Lobe-HOMO Lewis bases: HO–, H2O:, etc.
π-HOMO Lewis bases: allyl anion, ethene, etc.
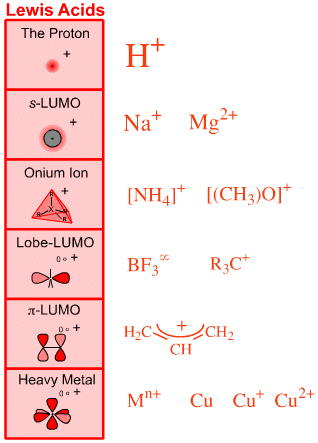
The previous page also introduced the notion that there are six distinct types of Lewis acid, again where classification is based on FMO topology. These six types of Lewis acid are:
The Proton Lewis acid, H+
s-LUMO Lewis acids, Na+, Mg2+, etc.
Onium Ion Lewis Acids: [NH4]+, [(Me3)3O]+, etc.
Lobe-LUMO Lewis Acids: BF3, R3C+, etc.
π-LUMO Lewis Acids: allyl cation, etc.
Heavy Metal Lewis Acids: metals & cations, etc.
Lewis Acid/Base Complexes
Lewis acid/base interaction chemistry can be stated in two ways:
- Electron pair donor Lewis bases "react with" or "complex with" or "interact with" electron pair acceptor Lewis acids to give Lewis acid/base complexes.
- The highest occupied molecular orbital (HOMO) of a Lewis base "reacts with" or "complexes with" or "interacts with" the lowest unoccupied molecular orbital (LUMO) of a Lewis acid to give a Lewis acid base complex with a bonding molecular orbital. The contributions of +/– charge and orbital overlap is described by the Klopman equation, here.
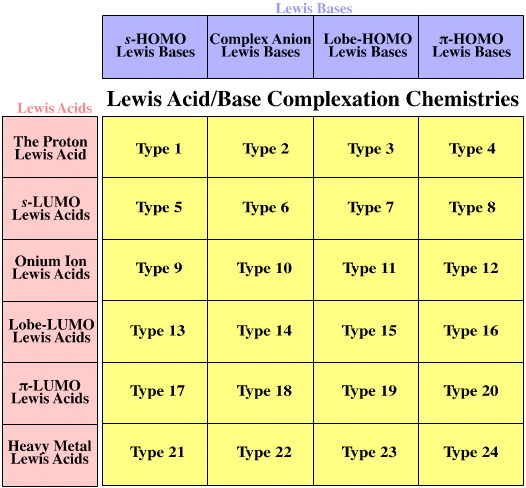
The six distinct types of Lewis acid and the four distinct types of Lewis base – where distinction is by frontier molecular orbital (FMO) topology, ie the shape, phase and geometry of the participating HOMOs and LUMOs – interact to give 24 distinct types of Lewis acid/base complex. This process can be visualised with the aid of the Lewis acid/base interaction matrix graphic:
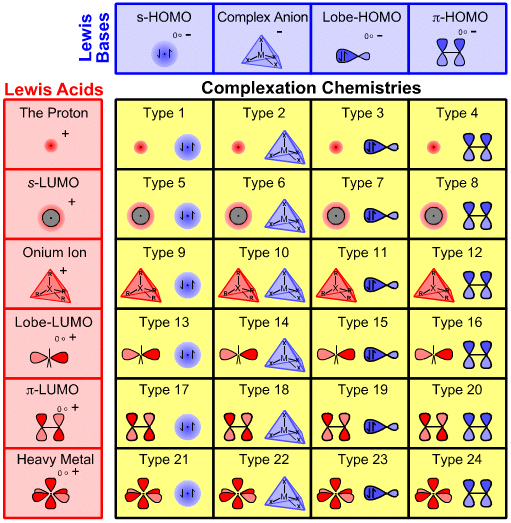
The Lewis acid/base interaction matrix – or interaction table, a type of Karnaugh map – has many, many properties. For example:
- Each cell of the Lewis acid/base interaction matrix contains distinct and characteristic chemistry.
- The matrix covers all Lewis acid/base reaction chemistry space.
We shall explore this object in some detail.
Across and Up-Down
The characteristic chemistry of a particular type of Lewis acid can be found by reading across the interaction matrix. Likewise, read up/down down for a particular type of Lewis base:
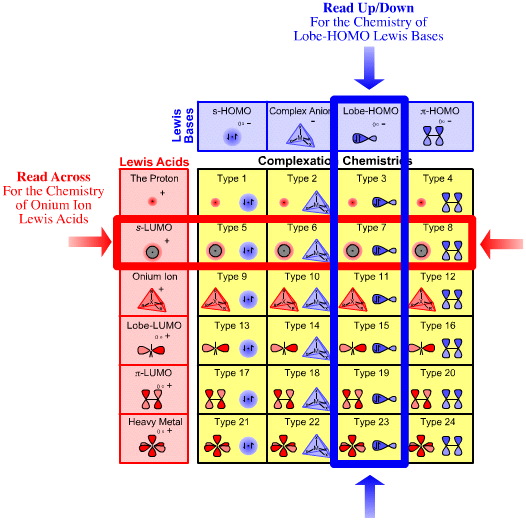
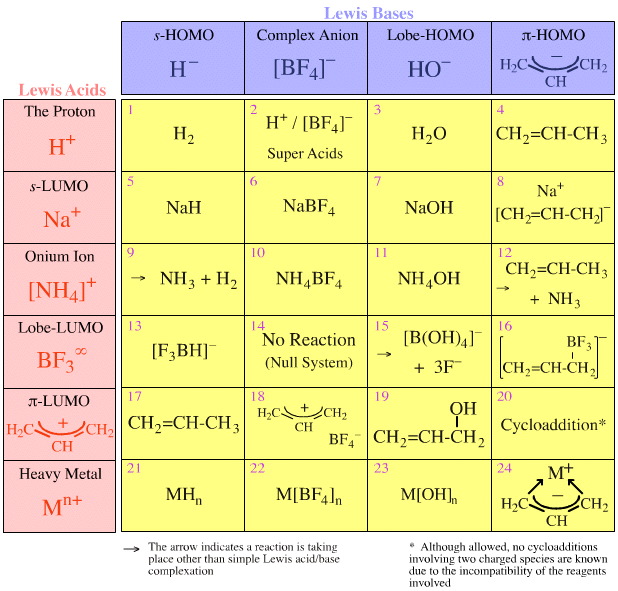
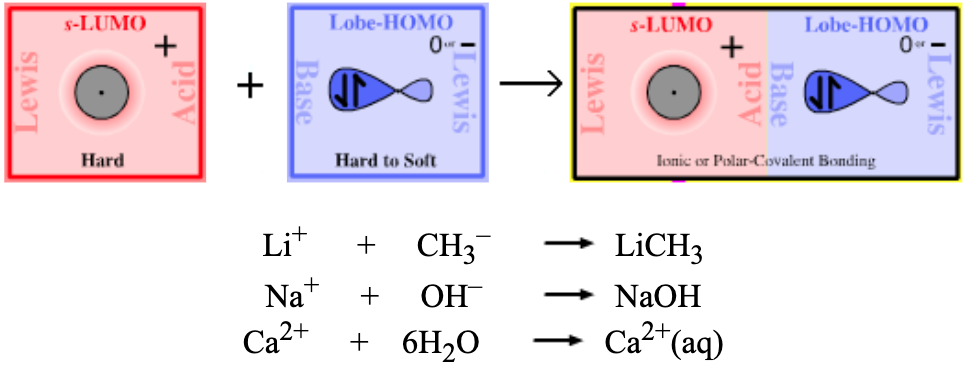
For example, an s-LUMO Lewis acid such as the sodium ion, Na+, interacts with Lobe-HOMO Lewis base such as the hydroxide ion, HO–, to give a Type 7 complex.
The point is that nearly all basic, proton abstracting reagents used in chemistry are also Type 7 complexes including:
methyl lithium, H3CLi
potassium hydroxide, KOH
sodium carbonate, Na2CO3
sodium hydrogen carbonate, NaHCO3
sodamide, NaNH2
lithium fluoride, LiF
calcium hydroxide, Ca(OH)2
sodium sulfide, Na2S
sodium cyanide, NaCN
magnesium oxide, MgO
barium sulfate, BaSO4
etc.
Complexation Type Numbers
Each of the interaction complex types is assigned a number from 1 to 24. These numbers are used to "keep track" and have no real significance... other than the fact that they are used in a self-consistent way in this webbook and The Chemical Thesaurus reaction chemistry database:
Real Species
When the schematic Lewis acid/base interaction matrix icons are replaced with real chemical species the nature and usefulness of the Lewis acid/base interaction matrix become apparent.
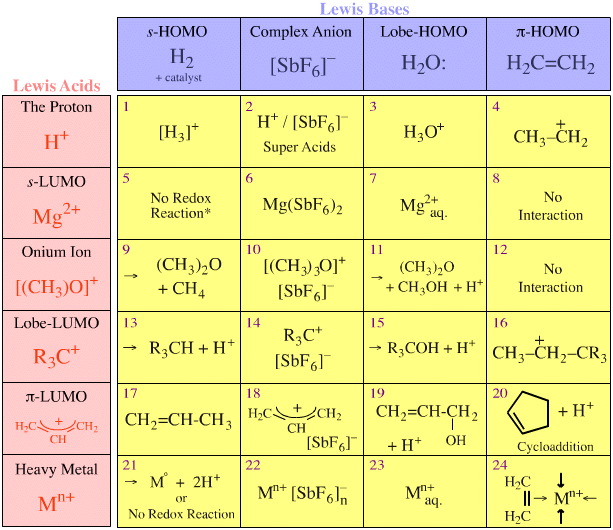
More Real Species
Many Lewis acid/base interactions initiate reaction mechanisms more involved than simple complexation. For example, the trimethyl oxonium ion reacts with water to give dimethyl ether and protonated methanol. This can be viewed as the transfer of a carbenium ion Lobe-LUMO Lewis acid from one Lobe-HOMO Lewis base to another. The interaction is an example of Type 11 Lewis acid/base reaction chemistry.
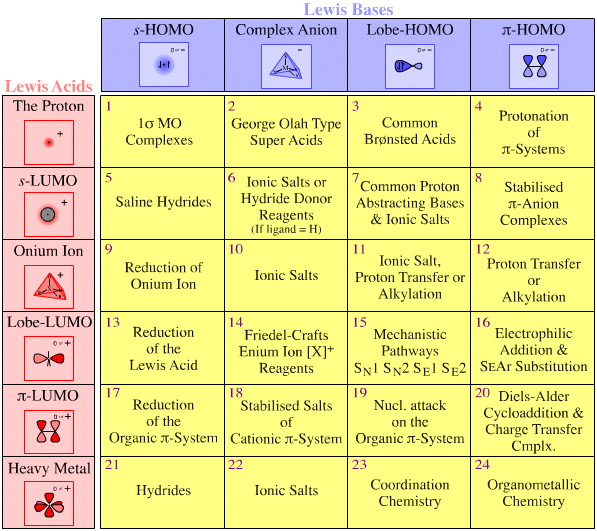
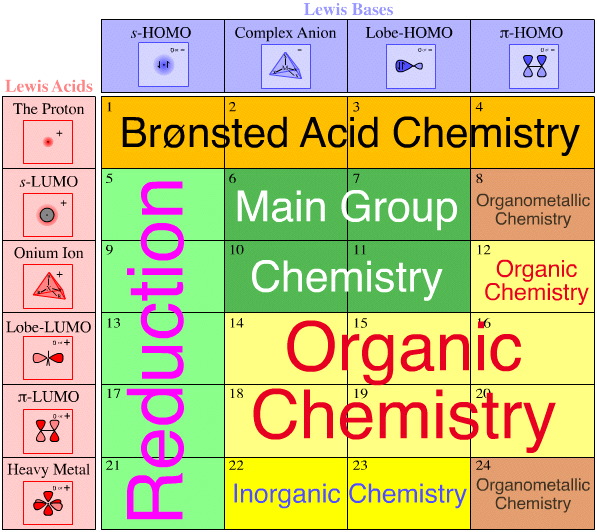
Lewis acid/Base Reaction Chemistries
Each of the 24 types of Lewis acid/base complexation can be mapped against well known types of reaction chemistry. For example, type 3 complexes are all "super acids" and Diels-Alder cycloaddition is associated with type 20 complexation.
Again, this logic is general in two ways:
• Firstly, each cell of the Lewis acid/base interaction matrix contains distinct and characteristic chemistry.
• Secondly, the matrix covers all of Lewis acid/base reaction chemistry space.
HSAB Analysis
In the 1960s, Ralph Pearson suggested that Lewis acids and Lewis bases should be classified as hard, borderline or soft, with the observation that: "Hard [Lewis] acids prefer to complex with hard [Lewis] bases and soft [Lewis] acids with soft [Lewis] bases", the HSAB principle (go here and here for more information).
The original HSAB analysis is very limited, but it regains its promise and power when applied after Lewis acids and Lewis bases are first classified by their frontier molecular orbital (FMO) topology. The analysis can now be used to describe the richness of bonding interactions:
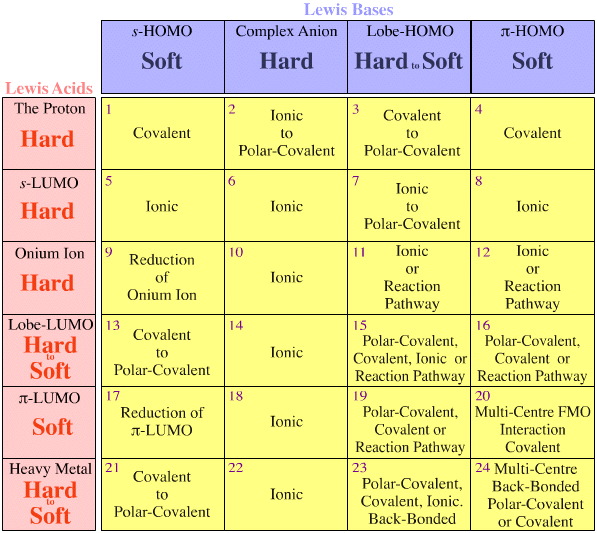
Traditional Areas of Chemistry
The Patterns in Reaction Chemistry analysis makes no initial distinction between the traditional organic, inorganic and organometallic reaction chemistries (divisions cause no end of confusion to students of the subject).
Yet these historical views can be mapped onto the Lewis acid/base interaction matrix.
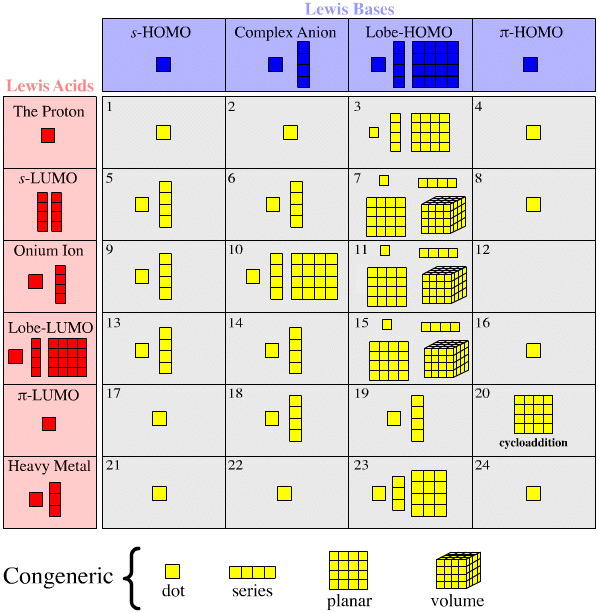
Searching for Congeneric Dots, Series, Planars and Volumes
Lewis acid and Lewis base types which are rich in congeneric arrays interact to give complex types which are rich in arrays.
Note, that on the diagram below there is not an exact one-to-one correspondence between the existence Lewis acid and Lewis base arrays and corresponding complex arrays. The reason is that not all complexation types are as interesting as each other. For example, Type 20 complexation is very rich... while Type 12 is not.
Two videos that develop the story in an audio-visual medium:
Introduction to the Lewis Acid/Base Interaction Matrix A Deeper Exploration into the Lewis Acid/Base Interaction Matrix, one cell at a time: |
The rest of this page is used to explore the 24 complexation chemistries in detail.
The reader may wish to come back to this page later and fast forward to species/species interaction page, here.
Type 1 Lewis Acid/Base Complexation Chemistry |
|
| 1σ2 Complexes | |
 |
|
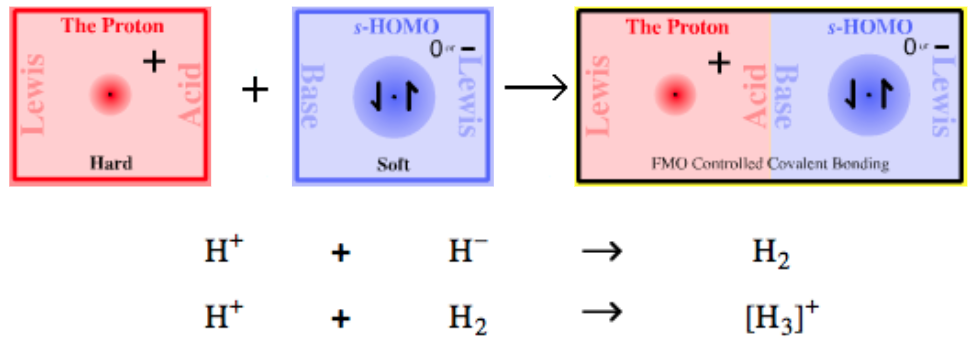
| Bonding: | Type 1 complexes, typified by hydrogen H2, are covalently bonded. The bonding is frontier molecular orbital (FMO) controlled. Hydrogen, H2, has a 1σ2 MO structure, ie they have two electrons in their 1σ molecular orbital. Find out more about the bonding diatomic species elsewhere in this webbook, here. |
| Charge: | Complexes can be neutral, H2, or positively charged [H3]+. |
| Chemistry: | Protons complex with hydride ions to form molecular hydrogen, H2, a uniquely simple and much studied diatomic molecule. The H+ + H– → H2 reaction is not reversible: H2 does not act as a proton donor (although at high temperature, when exposed to high energy UV radiation or when absorbed onto a metallic surface, H2 can homolytically dissociate: radical cleavage). As protons and hydride ions do not exist as independent species, they require "delivery" by donor complexes, ie reagents. Protons, H+ ions, are supplied by Brønsted acids and hydride ions by hydride donor complexes. For example, hydrogen chloride an H+ donor reacts with sodium hydride an H– donor to give diatomic hydrogen and sodium chloride: HCl + NaH → H2 + NaCl [H3]+, the product of H+ and H2, is the simplest possible triatomic molecular ion – it has only two electrons – and is of considerable theoretical interest.
(In this author's opinion the [H3]+ ion should be called the 'hydronium ion', and [OH3]+ should be the 'oxonium ion'.) |
| Congeneric Series: | Few series. |
Type 2 Lewis Acid/Base Complexation Chemistry |
|
| George Olah Superacids | |
 |
|
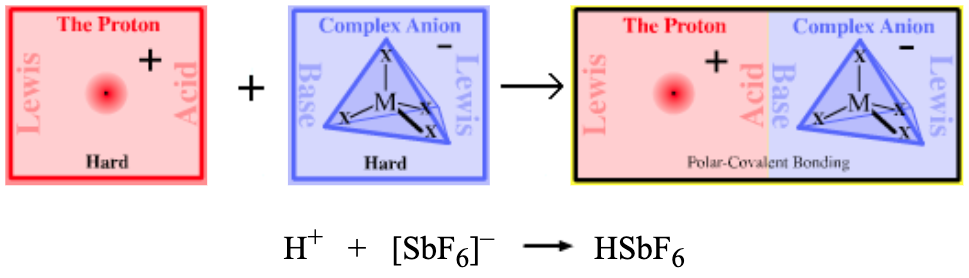
| Bonding: | Protons do not complex efficiently with complex anion Lewis bases as the proton must disrupt the anion’s high symmetry HOMO when a 1:1 complex forms. Hence, the resulting complex is a very powerful proton donor. Complexes are ionic/highly polar covalent. |
| Charge: | Superacids are neutral. |
| Chemistry: | Proton plus complex anion complexes must be prepared in ‘exotic’ solvents such as liquid SO2 and where the complex anion has fluoride ion ligands.
Such complexes are the strongest Brønsted acids known. Super [Brønsted] acids, 'superacids', have pKa values in the region -15 to -25, ie they are up to 20 orders of magnitude more [Brønsted] acidic than sulfuric acid. Superacids are able to fully protonate all Lewis base organic functional groups (as opposed to protonating a low equilibrium concentration). Alkenes, benzene, carbonyls and nitro functional groups are all protonated by superacids. For example: 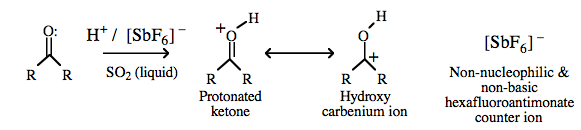
Read more on the Wikipedia superacid page. |
| Congeneric Series: | Few series. |
Type 3 Lewis Acid/Base Complexation Chemistry |
|
| Common Brønsted Acids | |
|
|
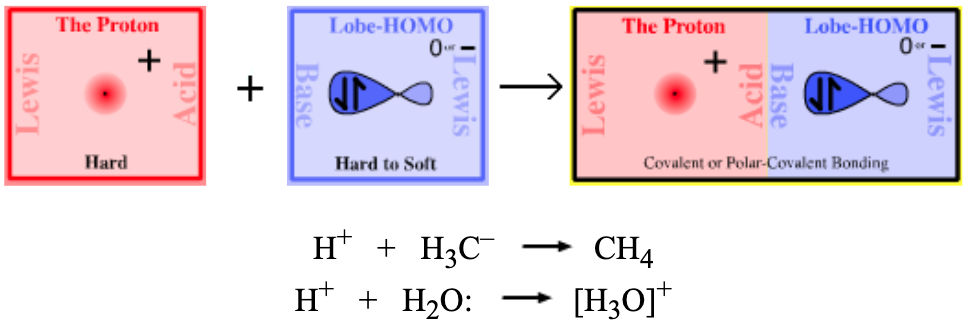
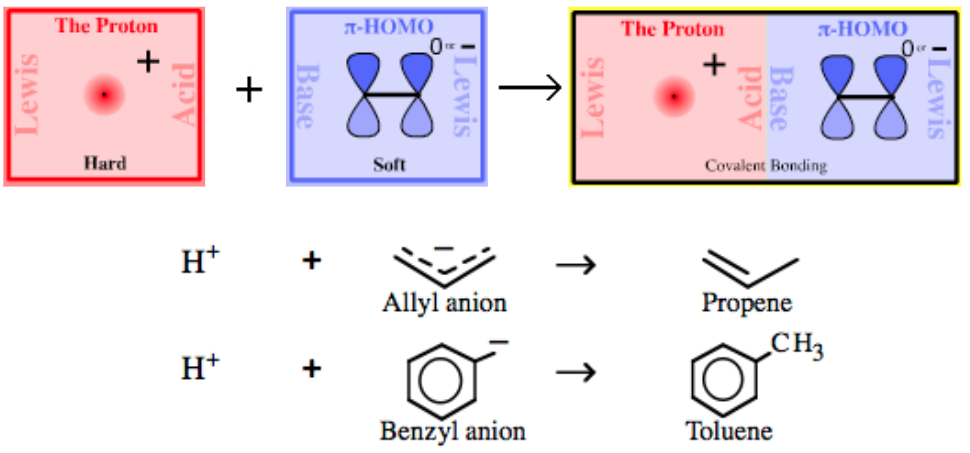
| Bonding: | The point-charge of the proton, H+, is able to efficiently penetrate and complex with the directional lobe shaped sp3/sp2/sp orbitals of all Lobe-HOMO Lewis bases. The resulting complexes are either covalent or polar-covalent. |
| Charge: | Complexes may be negatively charged, positively charged or they may be neutral. |
| Chemistry: | The vast majority of Brønsted acids are Type 3 Proton/Lobe-HOMO Lewis acid/base complexes, including all of the common mineral acids H2SO4, HCl, HNO3, H3PO4 etc. |
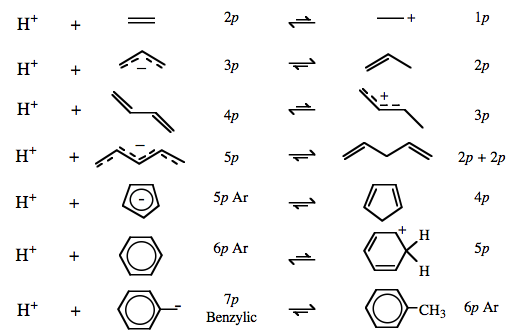
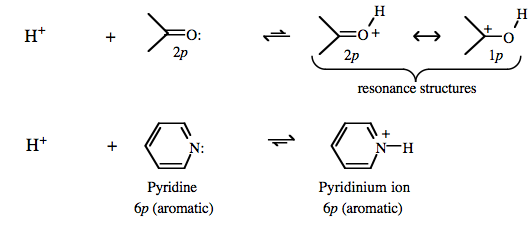
| Congeneric Series: | Complexes show regular Brønsted acid behavior across congeneric series, ie across and down the periodic table: 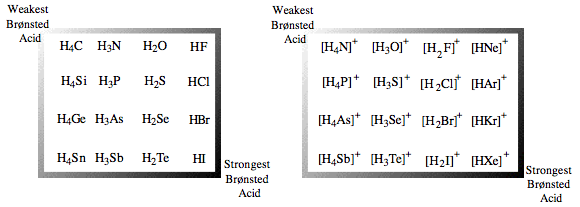 Brønsted acidity correlates with Lewis base to proton bond length, elsewhere in this webbook, here. CH4, methane and NH3, ammonia are the weakest Brønsted acids and have the shortest bond lengths while HI, hydrogen iodide and protonated xenon, [HXe]+, are the most acidic and have the longest bond lengths. Brønsted acidity correlates with Lewis base to proton bond length, elsewhere in this webbook, here. CH4, methane and NH3, ammonia are the weakest Brønsted acids and have the shortest bond lengths while HI, hydrogen iodide and protonated xenon, [HXe]+, are the most acidic and have the longest bond lengths.
Brønsted acidity increases with increasing conjugation and resulting π-stabilisation. 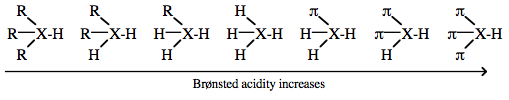
Read more elsewhere in this webbook, here. |
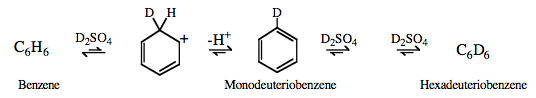
Type 5 Lewis Acid/Base Complexation Chemistry |
|
| Saline Hydrides | |
 |
|
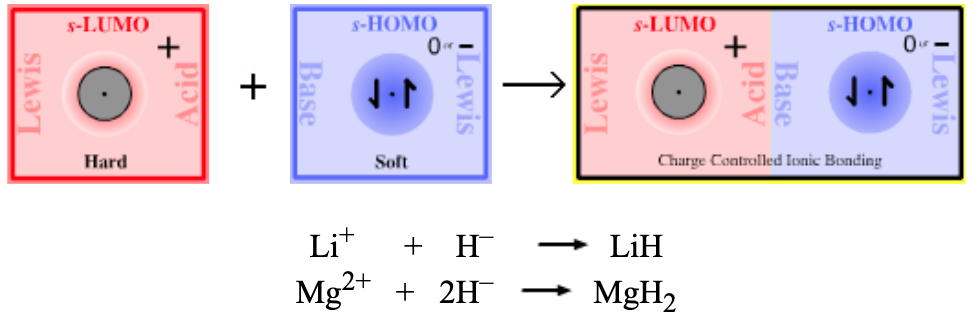
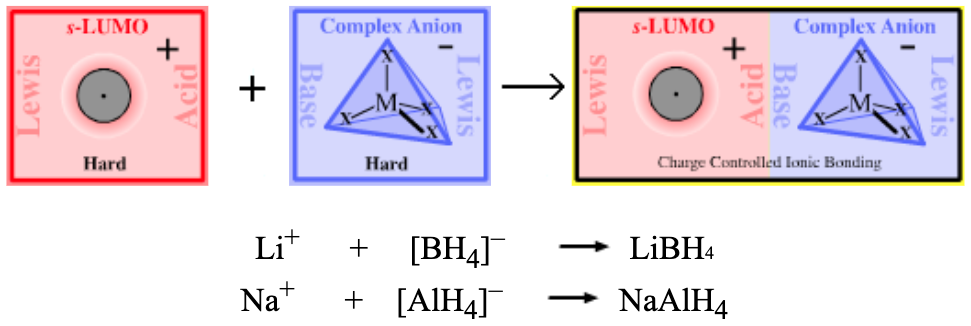
| Bonding: | Group I & II alkali and alkaline earth hydrides exist as ionic lattice solids. However, molecular 1:1 (LiH) and 1:2 (MgH2) complexes are formed in the vapor phase, although with difficulty as the compounds are liable to decompose back to elemental form. LiH is a much theoretically studied diatomic species. Studies show that bonding involves more than just s-LUMO/s-HOMO overlap. In valence bond (VB) terminology, the lithium’s 2s-LUMO mixes with (ie hybridizes with) a higher energy empty 2p orbital (ie the LUMO + 1 MO) to generate a directional sp hybrid bond. MO calculations show that the bonding in LiH involves 66% Li 2s/H1s overlap and 34% Li 2p/H 1s overlap. |
| Charge: | The charge on a Type 5 complex is always neutral. |
| Chemistry: | Saline hydride complexes either act as strong proton abstracting Bases or as donors of nucleophilic hydride ion. Organic chemists employ saline hydride complexes as proton abstracting Bases, with H2 being the conjugate Brønsted acid. Sodium hydride readily abstracts a proton from dimethyl sulfoxide (DMSO), pKa 35, to form sodium dimsyl: 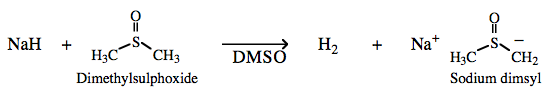
Inorganic chemists are more likely to use saline hydride reagents as a source of nucleophilic (and reducing) hydride ion, for example in the synthesis of sodium borohydride: 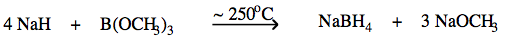 |
| Congeneric Series: | 
LiH NaH KH RbH CsH BeH2 MgH2 CaH2 SrH2 BaH2 |
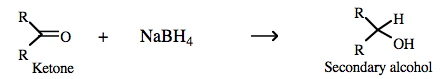
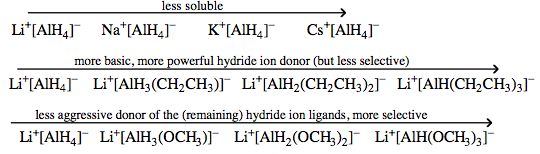
Type 7 Lewis Acid/Base Complexation Chemistry |
|
| Common Strong & Weak Brønsted Bases and Ionic Salts | |
 |
|
| Bonding: | The s-LUMO Lewis acids like to be multiply complexed by Lobe-HOMO Lewis base ligands to achieve maximum spherical symmetry about the cation. Complexes form as ionic crystal lattice (cesium fluoride) or they may generate polar-covalent bonds (methyl lithium). |
| Charge: | The charge on a Type 7 complex can be negative, neutral or positive. |
| Chemistry: | With a few important exceptions – for example, amines and the saline hydrides – most of the proton abstracting (Brønsted base) reagents are Type 7 Lewis acid/base complexes: NaOH KNH2 LiCH3 CH3COONa NaHCO3 Na2CO3 etc. Other Type 7 complexes, in which the Lobe-HOMO Lewis Base is the conjugate base of a strong Brønsted acid, are non-basic ionic salts: KCl NaI LiBr etc. In polar solvent solution (water, THF, DMF or DMSO), Type 7 complexes may show differing degrees of ion pairing: 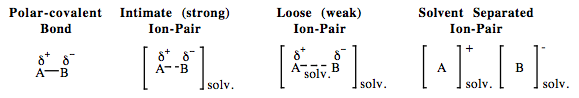
Polar solvation requires the solvent to have lone pair Lobe-HOMO centres which compete with the Type 7 Complexes’ Lobe-HOMO Lewis base centre. Methyl lithium, for example, is usually considered to be a 1:1 polarised covalent compound, however, in diethyl ether or THF solvents the lithium cation is co-complexed with solvent molecules. In alkane solvents alkyl lithium reagents aggregate into hexamers and higher structures, Wikipedia. |
| Congeneric Series: | There are many important congeneric series: alkyl carbanion reagents become more basic as the alkyl groups are replaced by hydrogen ligands, and as the metal counter ion becomes congenerically heavier. Regular changes in physical properties occur as pairs of congeneric Lewis acid/base series interact to form congeneric series, planars and volumes of complexes: 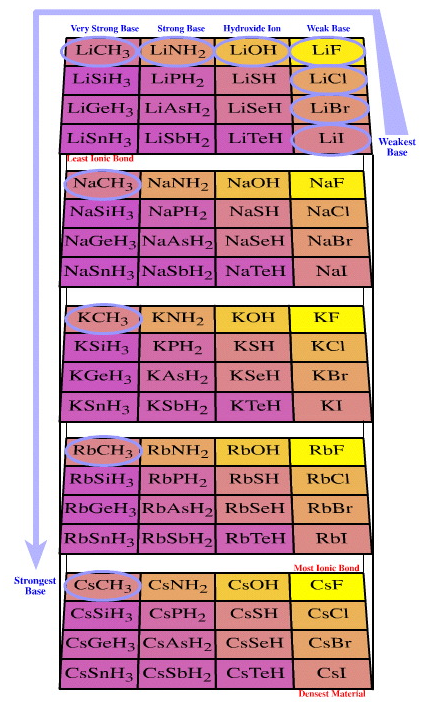 |
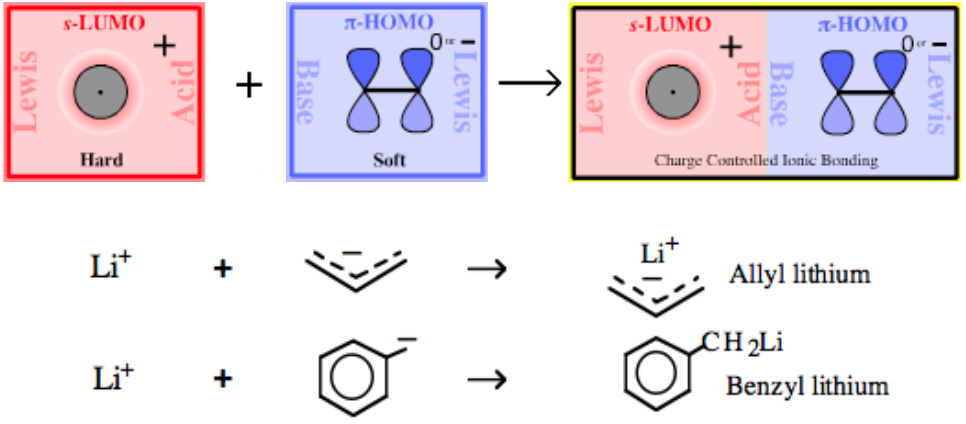
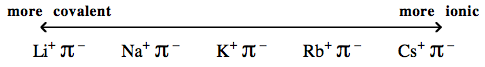
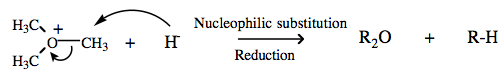
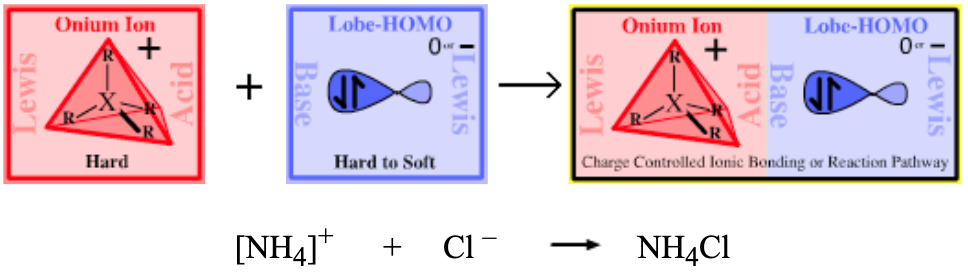
Type 12 Lewis Acid/Base Complexation Chemistry |
|
| Proton Transfer or Alkylation | |
 |
|
| Bonding: | Charge controlled ionic bonding, but... a proton transfer reaction is likely to occur rather than the formation of a complex. |
| Charge: | A reaction always occurs, rather than forming a Lewis acid/base complex so the idea of a charge on a complex is not applicable. |
| Chemistry: | Onium ions generally do not often form complexes with π-HOMO Lewis bases because H+ or R+ transfer (from the onium ion to the π-HOMO Lewis base) is likely to occur. The acetate ion can be dual classified as both as a Lobe-HOMO and π-HOMO Lewis base (the acetate ion is isoelectronic with the allyl anion). Thus, ammonium acetate can be considered to be a Type 12 complex: |
| Congeneric Series: | A reaction always occurs, rather than forming a Lewis acid/base complex so the idea of congeneric series is not applicable. |
Type 13 Lewis Acid/Base Complexation Chemistry |
|
| Reduction of Lewis Acid | |
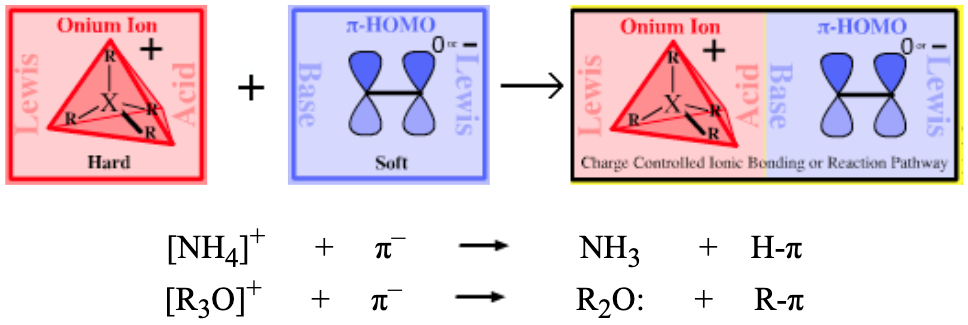
 |
|
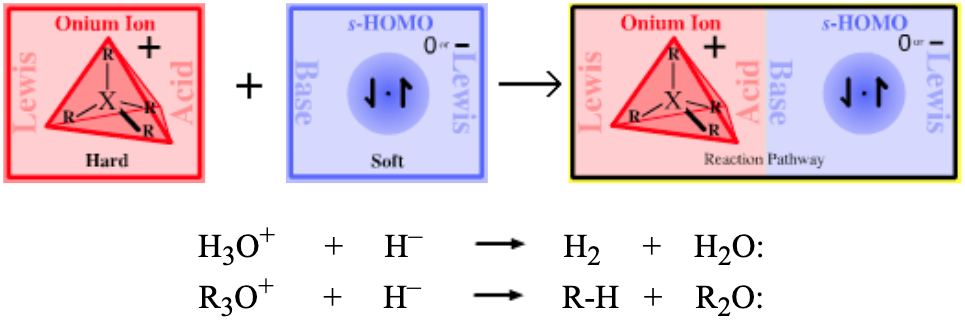
| Bonding: | s-HOMO Lewis bases are reducing agents, so complexation with hydride or hydrogen is also classed as reduction.
|
| Charge: | Complexes may be negatively charged or they may be neutral, or a reaction may occur. |
| Chemistry: | Carbenium ion (ie where the Lewis acid = R3C+ + hydride ion) complexation reactions are rare as both species interact strongly with their counter ions.
|
| Congeneric Series: | Few series. |
Type 14 Lewis Acid/Base Complexation Chemistry |
|
| Friedel-Crafts Reagents | |
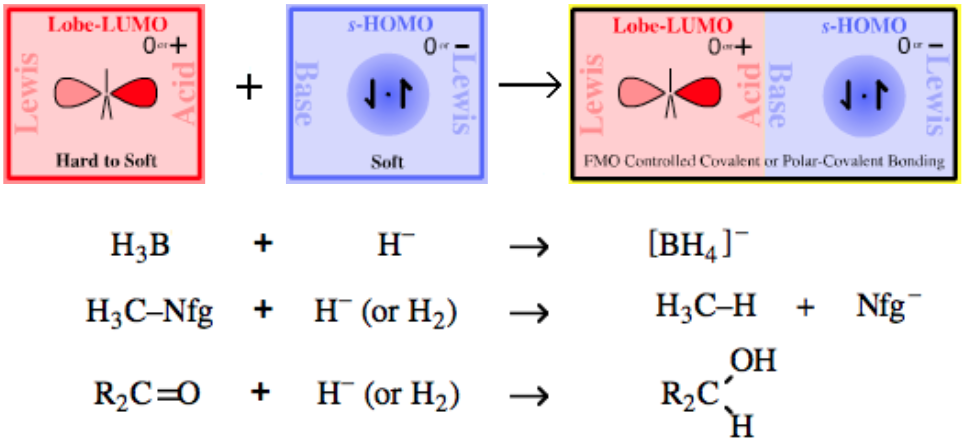
 |
|
| Bonding: | Charge controlled ionic complexes are highly reactive (transient) species which are prepared in solution where they show little or no ion pairing. |
| Charge: | The charge on a Type 14 complex is always neutral. |
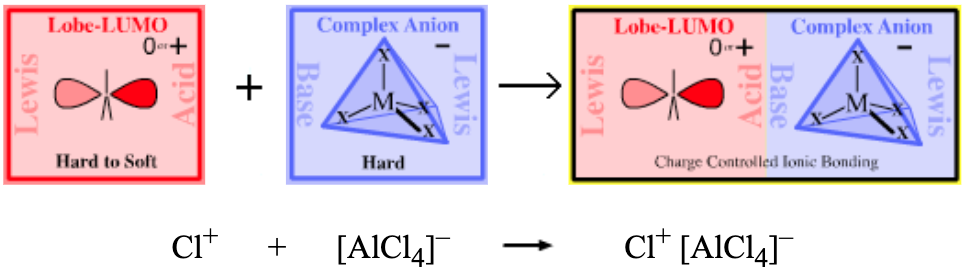
| Chemistry: | Type 14 complexes include electrophilic haloenium ion or carbenium ion reagents with non-electrophilic counter ions. Lobe-LUMO Lewis acids have a strong symbiotic affinity for halogen anion ligands, close in type to those they already possess. Thus:
Type 14 complexes are not prepared from the cation plus anion, but from the dihalogen, organic halide or acyl halide plus halophilic Lewis acid: Cl2 + AlCl3 → Cl+ [AlCl4]– R–Cl + AlCl3 → R+ [AlCl4]– The resulting Type 14 complexes act as sources of "naked" electrophilic Cl+, Br+ and R3C+, etc. that are able to undergo electrophilic aromatic substitution with benzene and other aromatics: 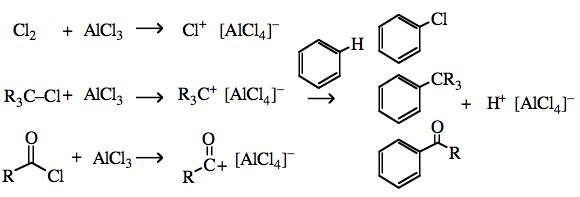 |
| Congeneric Series: | Few series. |
Type 15 Lewis Acid/Base Complexation Chemistry |
|
| Classical Organic Chemistry: Sn1, Sn2, SE1, SE2 Pathways | |
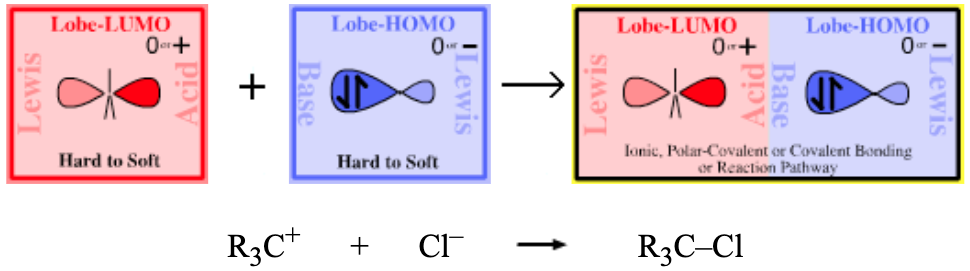 |
|
| Bonding: | The normal strong covalent and polarised covalent s-bonds of main group chemistry are generally Lobe-LUMO/Lobe-HOMO complexes: C-C C-N C-O C-F C-Br C-Nu C-Nfg Cl-Cl O-O S-Cl P-Br –B-N+ etc. In solution Lobe-LUMO/Lobe-HOMO complexes may be covalently bonded, polar-covalently bonded or strongly ion-paired, weakly ion-paired or solvent separated. |
| Charge: | Complexes may be negatively or positively charged or they may be neutral. |
| Chemistry: | Prochiral tertiary carbenium ions – carbenium ions with three different ligands and a solvent separated Lobe-HOMO Lewis Base counter ion – react with nucleophiles to give racemic mixtures of enantiomers: 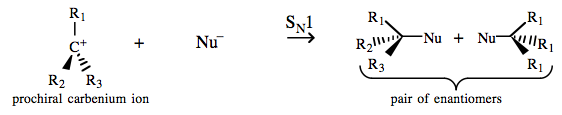
Chiral sp3 carbon/nucleofuge complexes react with nucleophiles via a concerted Sn2 mechanistic process in which the chiral centre inverts ‘like an umbrella’, a 'Walden inversion'. 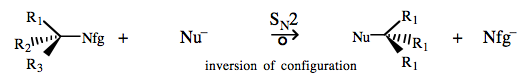
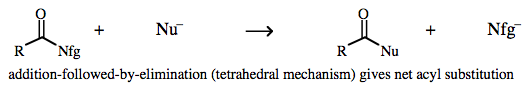
Strong ion-pairing makes concerted second order substitution more favorable than step-wise first order reactivity. Acyl chlorides and other carbonyl/Nfg complexes also undergo concerted nucleophilic substitution, although the mechanism is rather different:
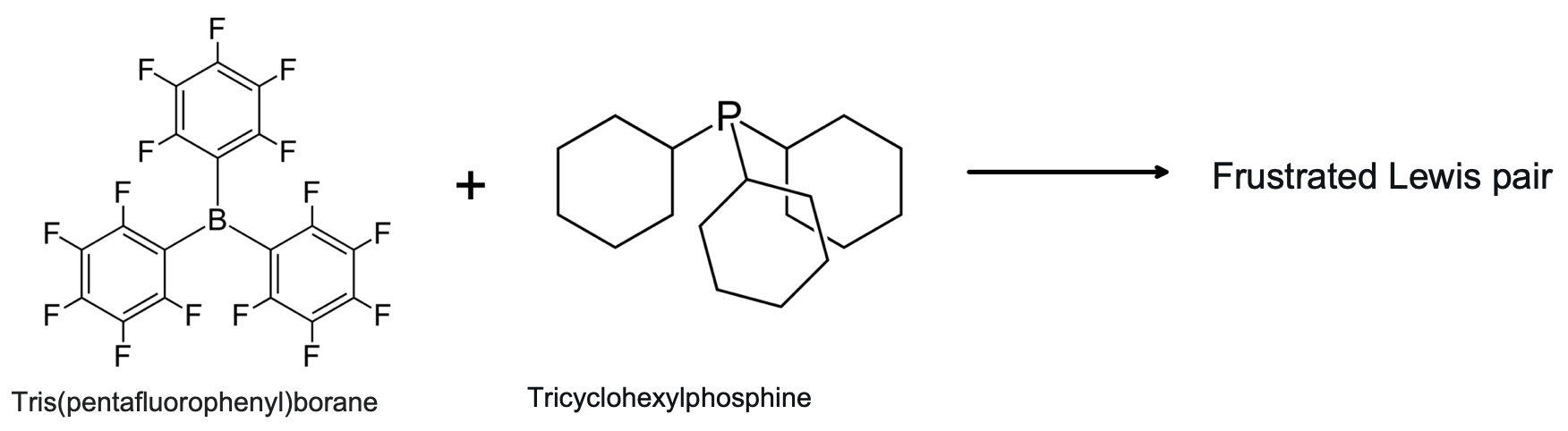
There is a type of Type 15 "non"-complex, the "Frustrated Lewis pair" where due to steric hinderance the Lewis acid/base adduct is unable to form. These species are synthetically useful in hydrogenation reactions, etc.:
|
| Congeneric Series: | There are many congeneric series and planars, for example: 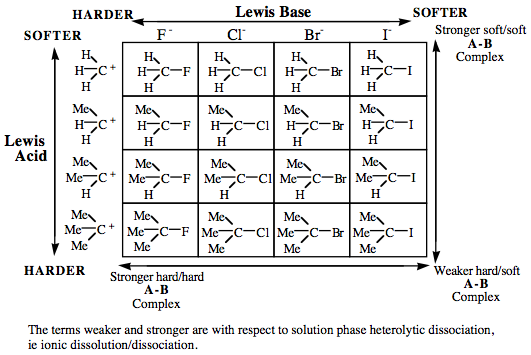 |
Type 16 Lewis Acid/Base Complexation Chemistry |
|
| Electrophilic Addition and SEAr Reactivity | |
|
|
| Bonding: | Halogen/alkene Type 16 complexes exhibit extensive back-bonding. |
| Charge: | Positively charged complex, such as the bromonium ion, or some reaction pathway may yield products with a variety of charges. |
| Chemistry: | Classical Organic Chemistry Many vacant p orbital Lobe-LUMO Lewis acids, particularly carbenium ions and acylium ions, are aggressive electrophiles, E+, able to react with π-HOMO organics, such as alkenes, via electrophilic addition or electrophilic addition-followed-by-elimination. For example: 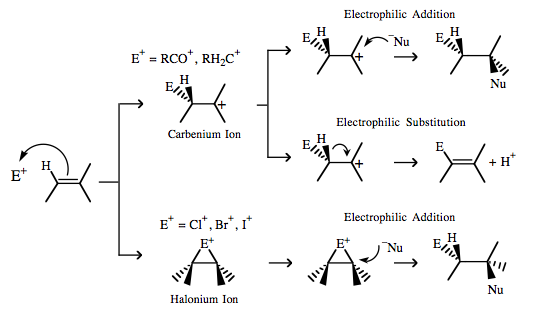
An electrophile, E+, may react with benzene and other aromatics to give (Friedel-Crafts or nitration) electrophilic aromatic substitution products: 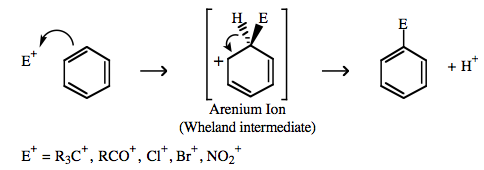 |
| Congeneric Series: | Few congeneric series or planars of interest. |
Type 17 Lewis Acid/Base Complexation Chemistry |
|
| Reduction of the Organic π-System | |
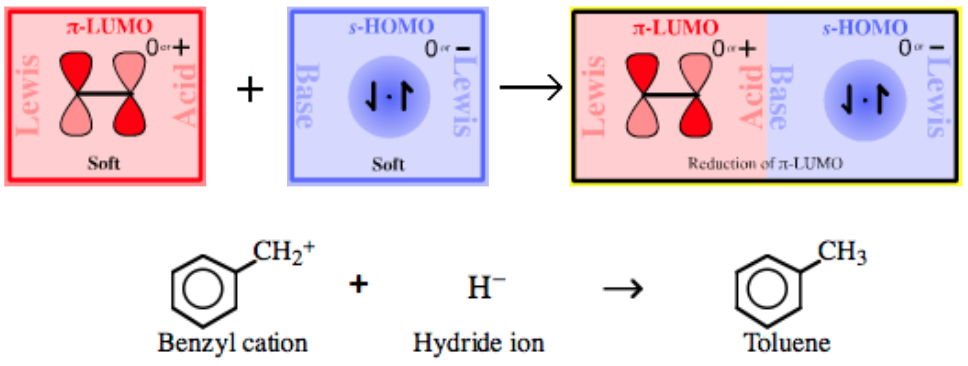 |
|
| Bonding: | π-System Lewis Acids form complexes with hydride ions which exist as C-H bonds. |
| Charge: | Complexes may be negatively charged or they may be neutral. |
| Chemistry: | π-System Lewis acids are reduced by s-HOMO Lewis bases: Hydride ion donor reagents, such as lithium aluminium hydride or sodium borohydride, both Type 6 complexes: 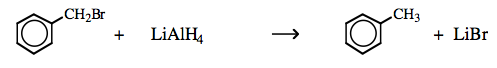
H2 plus transition metal catalyst: 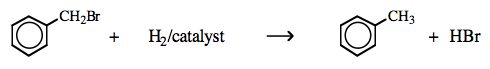
During such reductions the hydrogen is likely to be in the form of a transition metal/hydride Type 21 complex. The saline hydride reagents, NaH etc., Type 5 complexes, are generally too Brønsted basic to be used as organic reducing agents. |
| Congeneric Series: | Few series or planars of interest. |
Type 18 Lewis Acid/Base Complexation Chemistry |
|
| Cationic π-System Salts | |
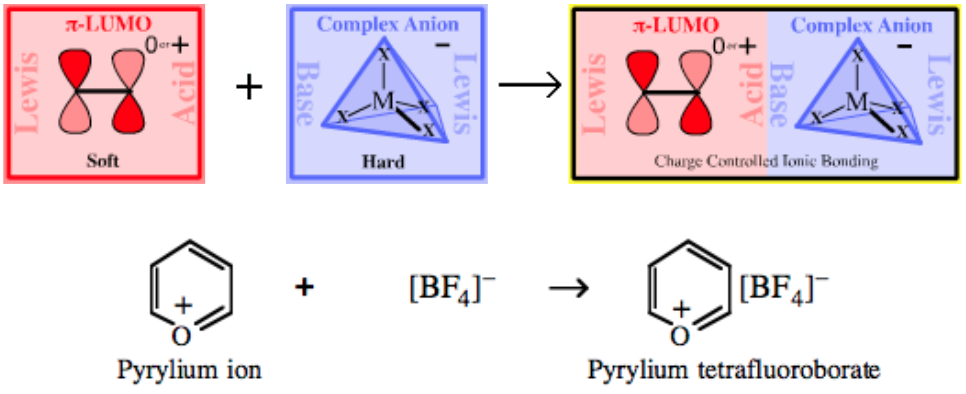 |
|
| Bonding: | Charge controlled ionic complexes which form solvent separated ions in polar solvents so making the interesting π-cations appear ‘naked’ for spectroscopic study.
|
| Charge: | Complexes are neutral. |
| Chemistry: | Exotic cationic π-systems, such as allyl & pentatrienyl cations: 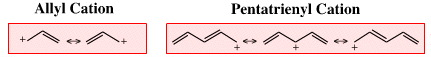
cyclopropenyl, cyclobutdienyl, tropylium & cyclooctatetrenely cations: 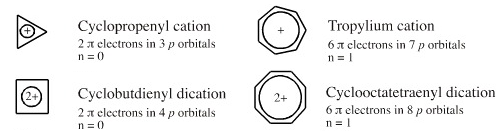
are reactive electrophilic entities that require very non-nucleophilic anionic counter ions, and tetrafluoroborate, [BF4]–, and hexafluorantimonate, [SbF6]–, type ions are ideal. |
| Congeneric Series: | Few of interest. |
Type 19 Lewis Acid/Base Complexation Chemistry |
|
| Nucleophilic Attack on π-Systems | |
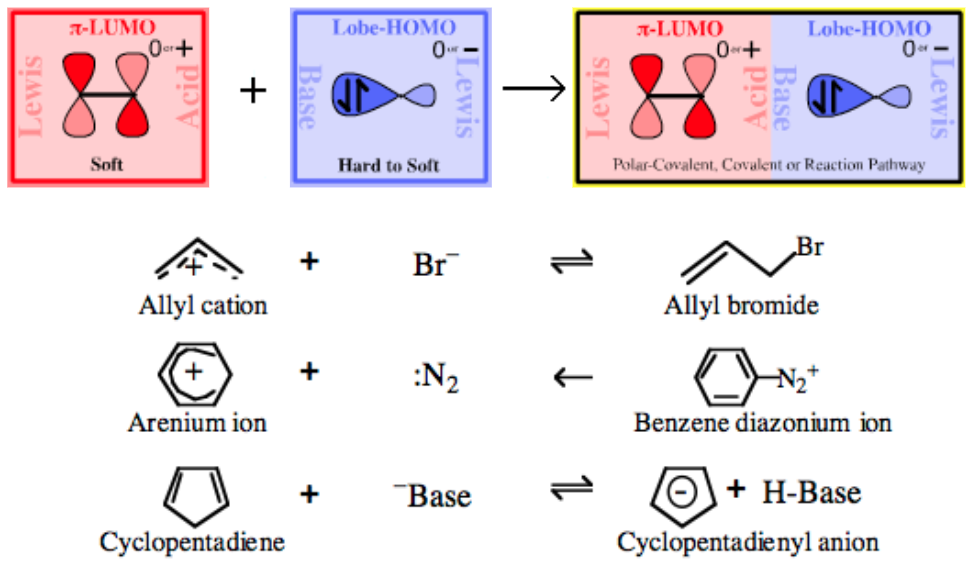 |
|
| Bonding: | Complexation leads to normal covalent and/or polar-covalent bonding. |
| Charge: | Complexes may be negatively charged, positively charged or they may be neutral. |
| Chemistry: | Nucleophilic attack, usually in an ambidentate manner, upon the π-LUMO Lewis Acid. One way to form π-LUMO Lewis Acids is to have a nucleofugal leaving group attached to a pro- π-LUMO Lewis Acid. Proton abstracters remove H+ to form the corresponding π-system. |
| Congeneric Series: | A number of interacting congeneric series are known, for example, phenyl methane carbenium ion/halogen anion complexes: 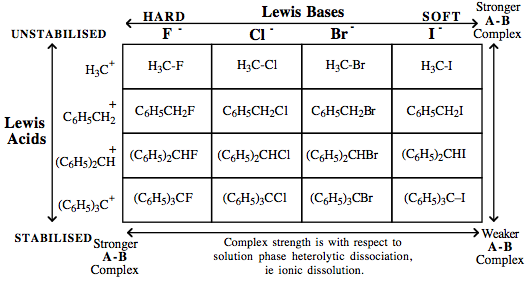 Chloride and other halogen anion nucleofuges are more easily substituted by hydroxide ion nucleophiles as –NO2 electron withdrawing group (EWG) functions are added (ortho and para) to the Nfg: Chloride and other halogen anion nucleofuges are more easily substituted by hydroxide ion nucleophiles as –NO2 electron withdrawing group (EWG) functions are added (ortho and para) to the Nfg: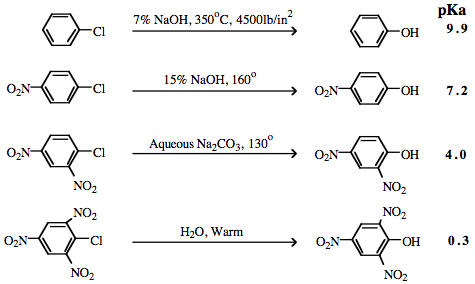 |
Type 20 Lewis Acid/Base Complexation Chemistry |
|
| π/π Interactions, including Diels-Alder Cycloaddition | |
 |
|
| Bonding: | If the π-LUMO Lewis acid and π-HOMO Lewis base species have suitable:
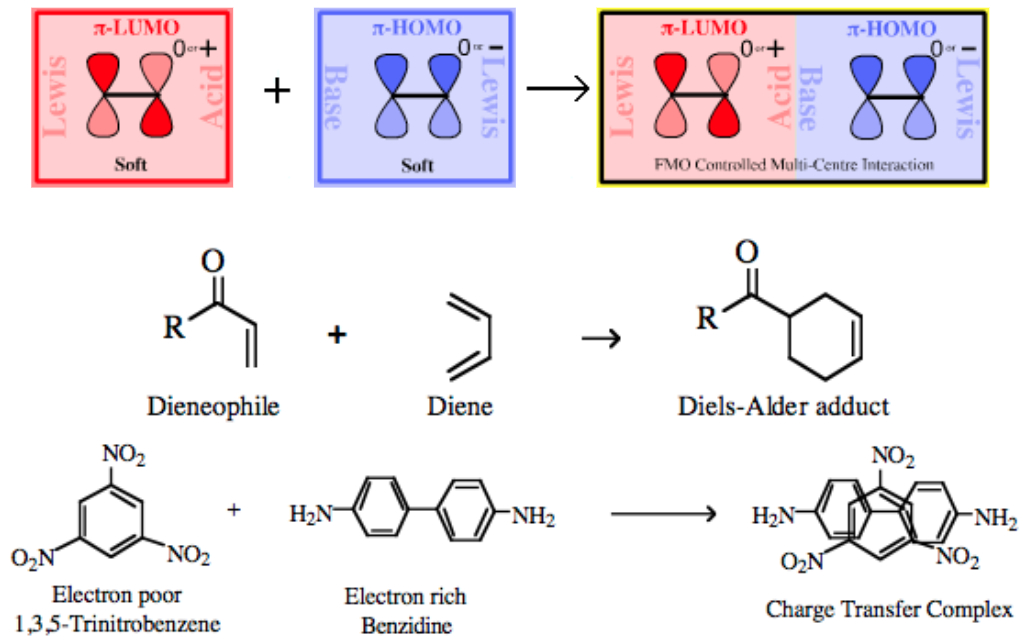
an orbital phase-symmetry controlled multi-centre π/π complexation reaction can take place with various the participating atoms rehybridizing to give a largely σ-bonded product. The transition state is deemed to proceed via a pericyclic intermediate, where "peri-" is a prefix meaning around or surrounding. The classic pericyclic interaction is the Diels-Alder cycloaddition between a diene and a dienophile: 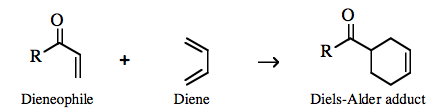
The orbital phase symmetry arguments required for cycloaddition to take place are interchangeable. Cycloaddition can either involve the HOMO of the diene + the LUMO of the dieneophile or the LUMO of the diene + the HOMO of the dieneophile: 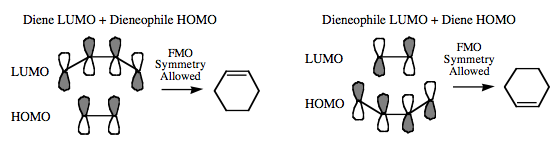
Pericyclic reactions are concerted: they take place in a single step. As a consequence, concerted processes provide allow for great Stereochemical control, and pericyclic processes are amongst the most useful of all synthetic methodologies available to the synthetic organic chemist.
Electron-poor δ+ π-systems can interact with electron-rich δ– π-systems. The initial attraction between is electrostatic (ionic) in nature to form a π/πcharge transfer complex. |
| Charge: | Complexes may be negatively charged, positively charged or they may be neutral. |
| Chemistry: | FMO Controlled Pericyclic Interactions Diels-Alder cycloaddition can be considered as multicentre π-LUMO plus π-HOMO Lewis acid/base Complexation chemistry. However, it is sometimes difficult to decide which species is acting as the Lewis acid and which is the Lewis base. In normal electron demand Diels-Alder cycloaddition chemistry the diene is electron rich, implying Lewis base character, and the alkene, the dieneophile, is electron deficient implying that the species is the Lewis acid: 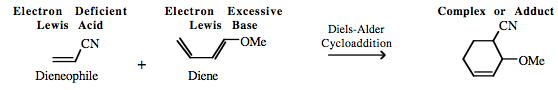
In reverse electron demand cycloaddition, the diene acts as the electron deficient Lewis acid and the dieneophile is the electron rich Lewis base: 
There are four general classes of pericyclic reaction process:
Charge Transfer Complexes π/π-Interactions can also lead to the formation of a charge-transfer complex, for example between the electron poor 1,3,5-trinitrobenzene and electron rich benzidine. 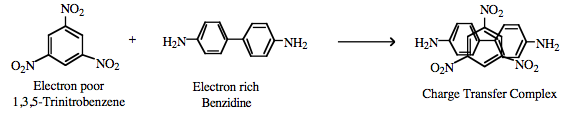
π/π-Interactions can also lead to the formation or conductive organic metals such as stacked TTF/TCNQ materials: 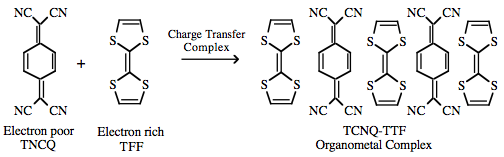 |
| Congeneric Series: | Few series. |
Type 21 Lewis Acid/Base Complexation Chemistry |
|
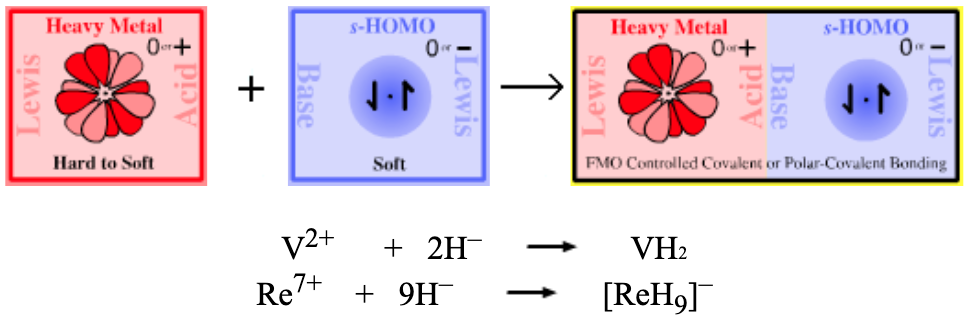
| Heavy Metal Hydrides | |
 |
|
| Bonding: | 
Metallic properties and non-stoichiometric compositions are common. The bonded hydrogen may be mobile. In various models the hydrogen may be present as H+ with the lost electrons going to the metal’s d-orbitals. In other models the hydrogen is assumed to have acquired electrons from the metallic conduction band and so be present as H–. Note that even though a metal may not form a hydride, it can still form a hydride complex ion, for example rhenium: Re7+ + 9H– → [ReH9]– |
| Charge: | Complexes may be negatively charged or they may be neutral. |
| Chemistry: | Hydride and hydrogen donors. Transition metal hydrides, MHn, can sometimes be formed by ‘dissolving’ hydrogen gas into a bulk metal to from a metal/hydride phase. This phase is the active ‘hydrogenating agent’ employed during catalytic hydrogenation. Palladium metal is able to absorb 900 times its own volume of hydrogen gas. Transition metal/hydride coordination complexes can be formed by nucleophilic displacement of a halogen (or other nucleofugal ligand) by hydride ions supplied by LiAlH4 or NaBH4: 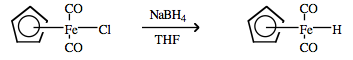
Metallic hydrides materials are usually dark powders or brittle solids. The bonding mechanism is important because heavy metal hydrides are being actively considered as hydrogen storage materials for hydrogen powered automobiles. |
| Congeneric Series: | Few series. |
Type 22 Lewis Acid/Base Complexation Chemistry |
|
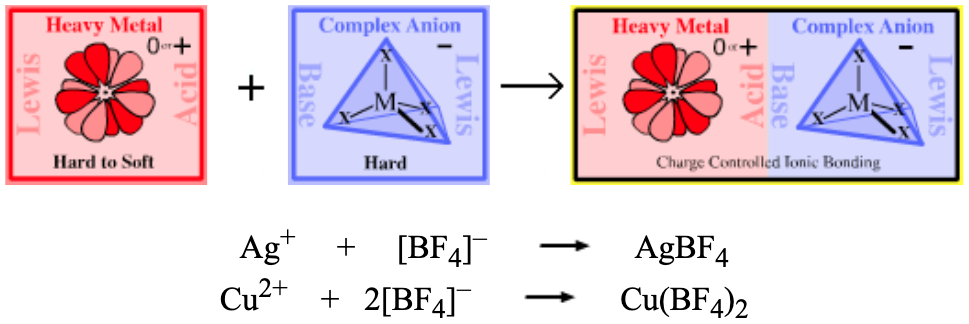
| Heavy Metal Ionic Salts | |
 |
|
| Bonding: | Compounds are charge-controlled ionic salts. In solution, ions are solvent separated. |
| Charge: | Complexes are neutral. |
| Chemistry: | There are few common heavy metal/complex anion complexes. That said, Pearson states in his early HSAB publications that transition metal ions of high oxidation state are harder than those of low oxidation state. Thus, we would only expect transition ionic metal/complex anion complexes to form with the harder higher oxidation state transition metal ions. This is what is found: the hard/hard copper(II) tetrafluoroborate complex, Cu[II] (BF4)2, is known, but the mixed soft/hard copper(I) tetrafluoroborate, Cu[I] BF4, is not. However, mixed soft/hard complex silver tetrafluoroborate, AgBF4, is known and is a useful chemical reagent. |
| Congeneric Series: | Few series. |
Type 23 Lewis Acid/Base Complexation Chemistry |
|
| Classical Inorganic Coordination Chemistry | |
 |
|
| Bonding: | The nature of bonding in heavy metal complexes, particularly transition metal complexes, is a vast subject with a long history and is of great technological importance: indeed the term ‘complex’ was first used to describe coordinated transition metal ions. The two most important bonding models are the electrostatic ligand-field theory and molecular orbital (MO) theory. Ligand-field theory assumes that the metal cation + anionic ligand interaction is primarily ionic. However, the more demanding MO model is computationally more accessible. The geometry of heavy metal complexes can usually be predicted by the valence shell electron pair repulsion VSEPR method, however, the effect of d and f orbitals must also be considered: [FeCl4]– is tetrahedral but the d8 complex [PdCl4]– is square planar. Transition metal complex ions may exhibit Jahn-Tellar distortion: Wikipedia, Robert J. Lancashire page & ScienceWorld. |
| Charge: | Complexes may be negatively charged, positively charged or they may be neutral. |
| Chemistry: | Lobe-HOMO Lewis bases form a multitude of complexes with heavy metal cations, indeed a great deal of classical inorganic coordination chemistry involves Type 23 complexation. Complexes may be linear, [AgCl2]–, tetrahedral, Ni(CO)4 or octahedral: 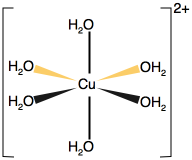
Heavy metals show multiple oxidation states due to loss of different numbers of d and f orbital electrons. Redox considerations can be as important as HOMO/LUMO interactions when predicting reactivity. Heavy metal complexes are usually coloured due to electronic transitions involving d or f orbitals and spectroscopic study can be used to probe the nature of the bonding in the complex. Sometimes small changes in ligands give rise to large spectral changes, and sometimes not. As a rule, d-d electronic transitions give rise to pale colors whereas charge-transfer transitions result in complexes with dark colours. Transition metals are employed in many biological systems where a protein molecule has a heavy metal ion at the active site, including: Hemoglobin, zinc finger protein & cytochrome: 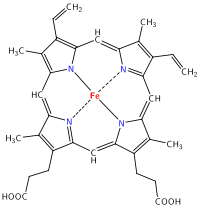
Structure of heme b 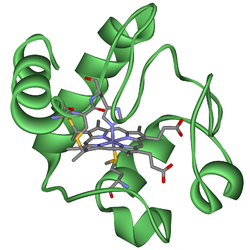
Cytochrome c with heme c. Industrial process catalysts may be heterogeneous (solid catalyst, liquid or gaseous reaction mixture) or homogeneous with respect to the reaction mixture. Wilkinson's catalyst, chlorotris(triphenylphosphine)rhodium(I), facilitates the homogeneous hydrogenation of alkenes: 
The phosphine ligands can be modified so as to give a chiral versions of Wilkinson's catalyst that are able to catalyse asymmetric hydrogenation. This approach has been developed into the Monsanto method for the production of L-DOPA, from Wikipedia: 
|
| Congeneric Series: | There are many congeneric series formed by ligand replacement. |
Type 24 Lewis Acid/Base Complexation Chemistry |
|||||||||||||||||||||||||||||||
| π-Organometallic Chemistry | |||||||||||||||||||||||||||||||
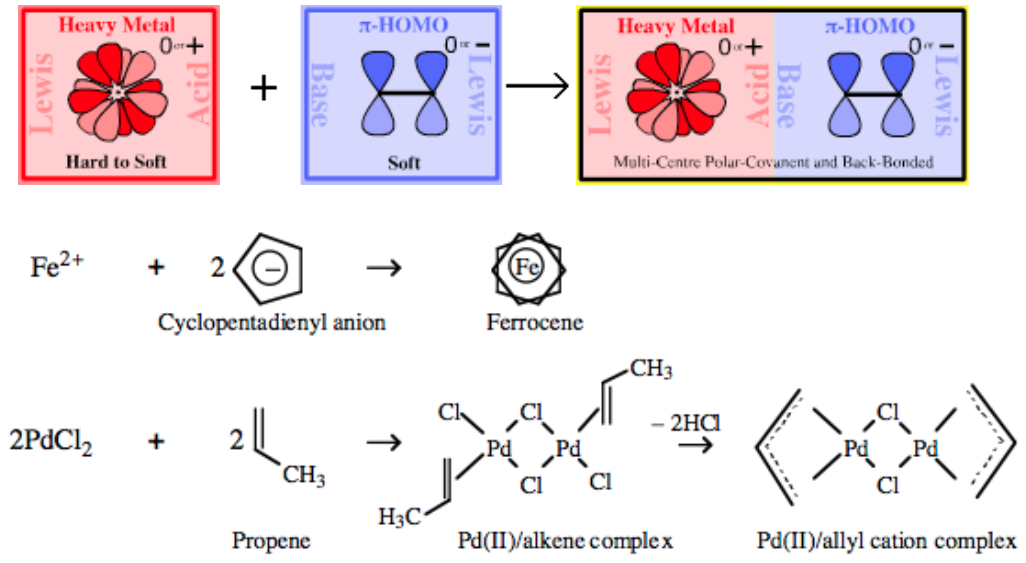 |
|||||||||||||||||||||||||||||||
| Bonding: | The bonding in π-organometallics must be considered using LCAO MO theory because the normal rules of valency break down. For example, is the valency of the Fe(II) ion in the metallocene in ferrocene, Fe(C5H5)2? Is the iron 2 valent or 10 valent? 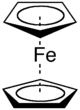
The hapto nomenclature, ηx, where the hapacity (Wikipedia) gives the number of conjugated p orbitals which ligate to a metal, rather than the number of electrons. |
||||||||||||||||||||||||||||||
| Charge: | Complexes may be negatively charged, positively charged or they may be neutral. | ||||||||||||||||||||||||||||||
| Chemistry: | There is a very extensive π-heavy metal chemistry known: all heavy metals are known to form π-organometallic compounds.
For example, chromium metallocenes: 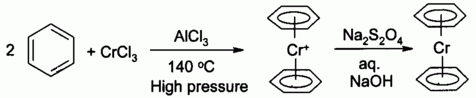
 |
||||||||||||||||||||||||||||||
| Congeneric Series: | Few of much interest. | ||||||||||||||||||||||||||||||
Hydrogen Bonding |
|||||||||
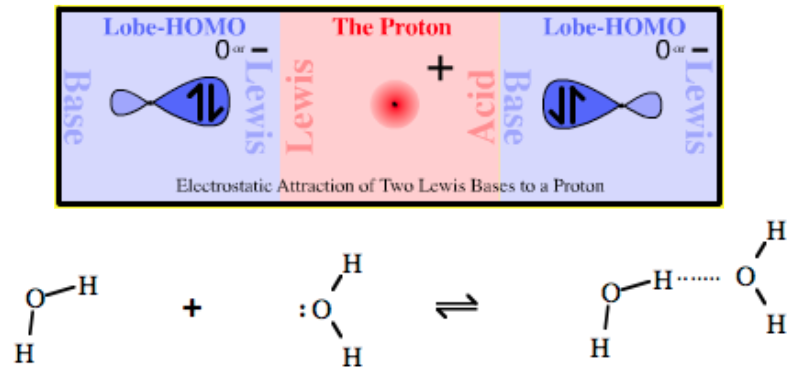
Proton Held Between Two Lewis Bases: Lewis Base/Proton/Lewis Base Complex |
|||||||||
 |
|||||||||
| Bonding: | Hydrogen bonding occurs when a proton Lewis acid, H+, is held between two Lewis bases. The hydrogen bond is a weak type of complexation deemed responsible for the high boiling points of water, alcohols, carboxylic acids etc. and the high solubility of (low molecular weight) alcohols, carboxylic acids and sugars in water. There is a long and detailed discussion on the Wikipedia hydrogen bond page, however, while this page gives lots of description and illustration it avoids an explanation of the true nature of hydrogen bonding. While hydrogen bonding does occur when a proton Lewis acid, H+, is held between two Lewis bases there a severe problem. The simple ‘Lewis base–proton–Lewis base’ model cannot exist on simple LACO MO grounds as there are too many electrons!
Lewis base-H+ bonds are generally highly polar structures and it is easy to consider the hydrogen bond to result from dipole-dipole electrostatic attraction. This is certainly the model and used by many text books. "The lone pair of electrons on one water's δ– oxygen attracts the δ+ hydrogen on an adjacent water molecule: 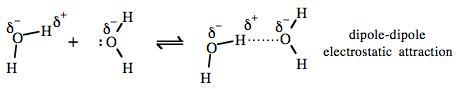
There is a clues to the true nature of the hydrogen bond, and it comes from reaction chemistry: All Brønsted acid proton transfer reactions pass through a hydrogen bonded intermediate transition state. Ammonia reacts with hydrogen chloride to produce ammonium chloride, NH4Cl. However, if the reaction is performed at -269°C, 4K, the NH3/HCl hydrogen bonded complex can be trapped – by matrix isolation – and studied by infrared vibrational spectroscopy. This experimental system shows there to be a structure in which the hydrogen atom rapidly moves, vibrates, between the chloride and amine Lewis base centres: 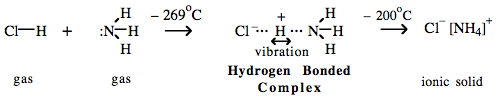
When the proton vibrates in the hydrogen bond it moves from a state in which it is bonded to one Lewis base to a state in which it is bonded to the other: 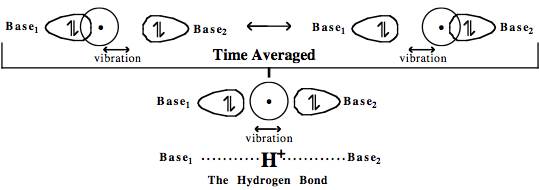
The time averaged effect, the superposition, is for the two Lewis bases to be attracted to each other through the hydrogen atom.
But why?
|
||||||||
| Charge: | Neutral. | ||||||||
| Chemistry: | Water, oxygen hydride, is a liquid at room temperature yet all of the other main group hydrides close to water in the periodic table are gases at 25°C and 1.0 atm pressure:
While NH3 and HF do exhibit hydrogen bonding and elevated boiling points, it seems that H2O is ideally suited to exhibit hydrogen bonding. Another strange manifestation of hydrogen bonding is that water has its maximum density at 4°C. Thus, water ice is less dense than liquid water and it floats. Most solids have greater density than the liquid. Crystal structure of hexagonal ice. Gray dashed lines indicate hydrogen bonds (Wikipedia): 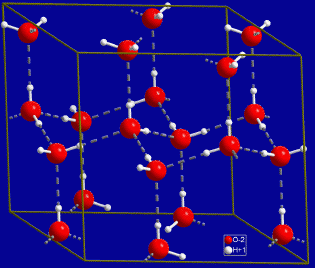
Water hydrogen bonds with ammonia, and either molecule can behave as the H+ donor or acceptor. More complicated molecules can have different types of hydrogen bonding function (from Wikipedia): 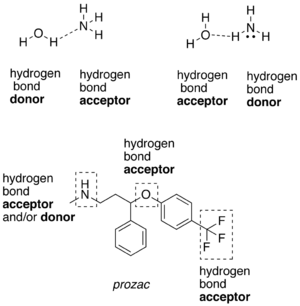
Hydrogen bonding is seen with all molecules possessing -OH functions, including alcohols, carboxylic acids and sugars such as glucose. Carboxylic acids such as acetic acid exist as gas phase dimers: 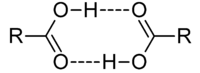
β-Diketones partially exist the hydrogen bonded cyclic-enol form: 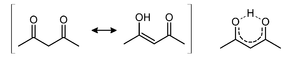
Hydrogen bonding is of immense importance in molecular biology as it constitutes the glue which holds together the twin strands of the DNA double helix and is responsible for secondary, alpha–helix & beta-sheet, and tertiary protein structure. Side view of an α-helix of alanine residues in atomic detail. Two hydrogen bonds to the same peptide group are highlighted in magenta: 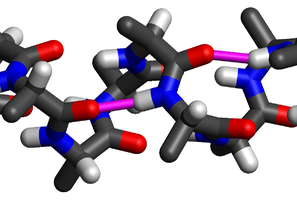
Diagram of a section of β-pleated sheet with H-bonding between protein strands: 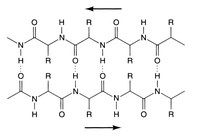
Chemical structure of DNA. Hydrogen bonds between A=T and between C≡G are shown as dotted lines: 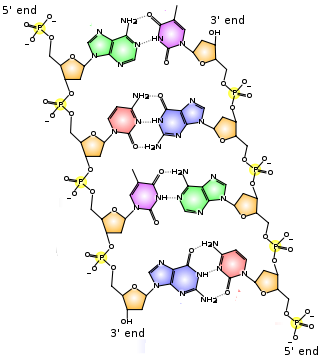 |
||||||||
| Congeneric Series: | Hydrogen bonding can be studied by substituting D+ for H+, but the congeneric series concept is not really very useful. | ||||||||
Three-Center Two-Electron Bridge Bonding Ligands |
|
| Lewis Acid–Lewis Base–Lewis Acid Complex | |
|
|
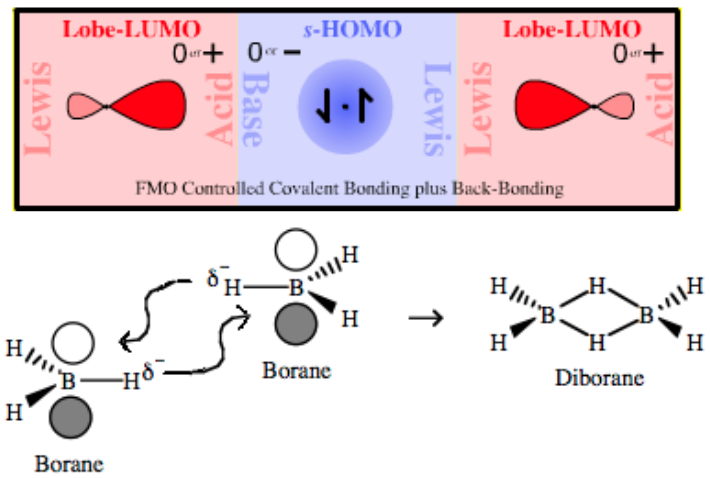
| Bonding: | A 3-center-2-electron bond, 3c-2e, is an electron deficient chemical bond where three atoms share two electrons, Wikipedia. A bridging ligand, Wikipedia, is a ligand that connects two or more atoms, usually metal ions. The ligand may be atomic or polyatomic. Virtually all complex organic compounds can serve as bridging ligands, so the term is usually restricted to small ligands such as hydride, halide or pseudohalides or to ligands linking two metals. Bridge bonding occurs when a Lewis base is held between a pair of vacant p or d orbital Lewis Acids. The system has two electrons which lead to the formation of a single bonding MO. 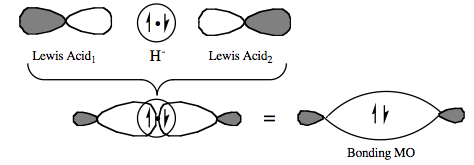
Borane, BH3, does not exist at room temperature because it dimerises to diborane. Hydride bridging bonds are found in diborane, B2H6., where the two central hydrogen atoms are simultaneously bonded to both boron atoms in 3c-2e bonds, Wikipedia: The compound commonly called "trimethylaluminium, Al(CH3)3," is actually the dimer with the formula Al2(CH3)6, Wikipedia: 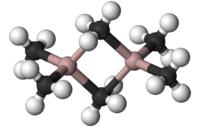 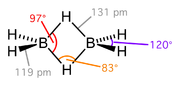
Halogen anion bridging bonds as in palladium[II] chloride, PdCl2: 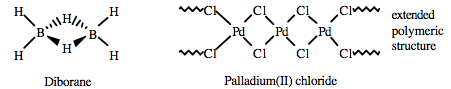
In the ruthenium complex, (η6-C6H6)2Ru2Cl2(μ-Cl)2, two chloride ligands are terminal and two are μ2 bridging. The η in the beginning of the formula denotes the hapticity of the benzene ligands, Wikipedia: 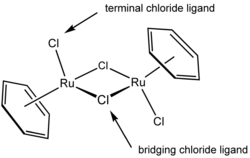
Virtually all ligands are known to bridge, with the exception of amines and ammonia. Particularly common inorganic bridging ligands, from Wikipedia, are:
|
| Charge: | No charge. |
| Chemistry: | Many of these reactive reagents behave as if their structures are the simple molecular lobe LUMO Lewis acids: BH3 and Al(CH3)3. |
| Congeneric Series: | The congeneric series concept is not really very useful here. |
van der Waals Complexation |
|||||||||||||||||
| Lewis Base–Proton–Lewis Base Complex | |||||||||||||||||
|
|||||||||||||||||
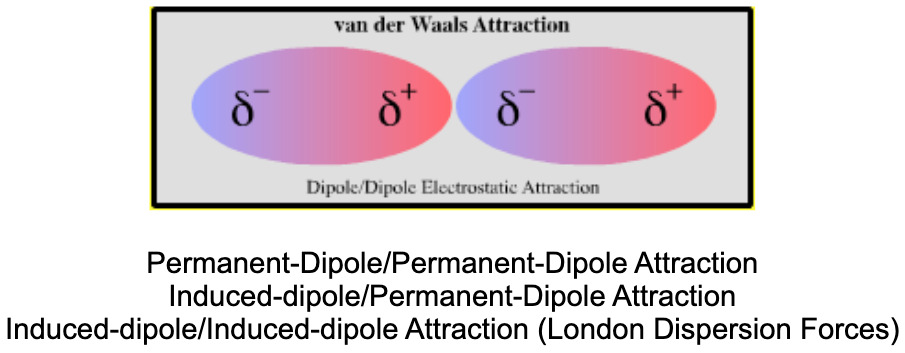
| Bonding: | There are several types of van der Waals attraction:
It is tempting to consider these forces to be of different strengths, but it is the distance range that is more important. Dipole/dipole attraction is relatively long range in action while the London spontaneous-dipole/Induced-dipole attraction requires contact between the van der Waals surfaces: the molecules need to touch.
Permanent-Dipole/Permanent-Dipole Attraction: Molecules with permanent dipole moments, polar molecules, such as iodine chloride, ICl, exhibit dipole-dipole attraction. The iodine end of iodine chloride is δ+ and the chlorine end is δ–. Molecules interact with each other so that the dipoles line up end-to-end: 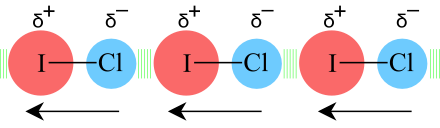
All molecules with a permanent dipole exhibit permanent-dipole/permanent-dipole attraction. At temperatures below the material’s melting point, the structure will show long range order and crystallinity.
Permanent-Dipole/Induced-Dipole Attraction : Molecular dipoles (polar molecules) are able to induce weak dipoles in adjacent non-polar species. The effect gives rise to a weaker attraction than dipole-dipole attraction. 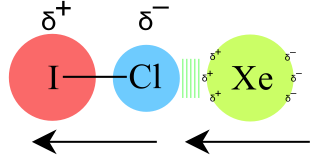
London Dispersion Force (LDF) Attraction: The very fact that it is possible to liquefy helium – and indeed all molecular materials – demonstrates that there must be some type of inter-molecular attraction taking place between the helium atoms. (Helium is a molecular material, where the helium molecule consists of just one atom.) The attraction is known as the London dispersion force and is deemed to arise from short time scale fluctuations in the electronic structure of species which results in the formation of instantaneous dipoles. The instantaneous-induced-dipole/induced-dipole London dispersion forces (LDF) are surprisingly strong but they only act at very short range. It is as if the surface of even neutral, non-polar molecules like methane, CH4, are 'sticky'.
All molecules exhibit London dispersion forces and the strength increases with the size/surface area of the molecule. This logic can be used to explains the increasing boiling and sublimation temperatures of the halogens. Going down the periodic table the atoms become larger, so the diatomic molecules become larger and their surface area becomes larger. Thus, the van der Waals forces increase and so do the boiling points: F2 < Cl2 < Br2 < I2 Likewise, longer chain alkanes have higher boiling points than shorter chain alkanes. Branching, which decreases surface area, reduces boiling point. Which is stronger: dipole/dipole or London forces?
Gecko Toes, Setae and van der Waals Forces: "The toes of the gecko have developed a special adaptation that allows them to adhere to most surfaces. Recent studies of the spatula tipped setae on gecko footpads demonstrate that the attractive forces that hold geckos to surfaces are van der Waals interactions between the finely divided setae and the surfaces themselves. Every square millimeter of a gecko's footpad contains about 14,000 hair-like setae." Wikipedia:  |
||||||||||||||||
| Charge: | No charge. | ||||||||||||||||
| Chemistry: | As well as giving an explanation of the forces involved in the melting, sublimation and boiling of molecular materials, van der Waals forces can help explain dissolution of a solute in a solvent. As a first approximation: "Like Dissolves Like":
|
||||||||||||||||
| Congeneric Series: | The congeneric series concept is not useful here. | ||||||||||||||||
Guest-Host Complexation |
|
| Molecular Shape Recognition Complex | |
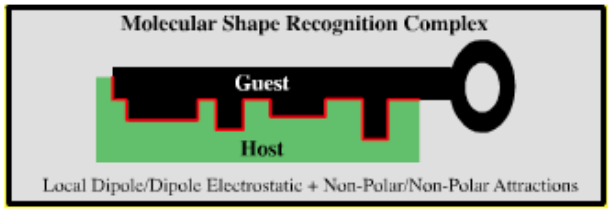 |
|
| Bonding: | Guest-Host Complexation "Complexes composed of two or more molecules or ions held together in unique structural relationships by hydrogen bonding, ion pairing or by van der Waals force, but not by full covalent bonds." Wikipedia, p-xylylenediammonium bound within a cucurbituril: 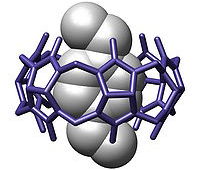
Molecular Recognition "The term molecular recognition refers to the specific interaction between two or more molecules through non covalent bonding such as hydrogen bonding, metal coordination, hydrophobic forces, van der Waals forces, pi-pi interactions, electrostatic and/or electromagnetic effects. The host and guest involved in molecular recognition exhibit molecular complementarity." Wikipedia
Inclusion Compounds "An inclusion compound is a complexing which one chemical compound, the host, forms a cavity in which molecules of a second guest compound are located. The definition of inclusion compounds is very broad, extending to channels formed between molecules in a crystal lattice in which guest molecules can fit. If the spaces in the host lattice are enclosed on all sides so that the guest species is ‘trapped’ as in a cage, the compound is known as a clathrate. In molecular encapsulation a guest molecule is actually trapped inside another molecule." Wikipedia Solid urea, (NH2)2C=O, can accommodate octane or 1-bromoctane, but not 2-methyl heptane or 2-bromooctane, as guest molecules in a hexagonal host lattice structure which contains long guest channels about 500pm in diameter. Zeolites are aluminosilicate minerals (natural and synthetic) which have open structures able to accommodate a wide range of guest molecules. The microporous molecular structure of a zeolite ZSM-5: 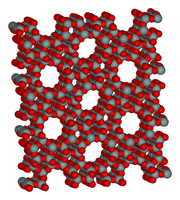
Biological Systems Molecular recognition plays an important role in biological systems and is observed in between receptor-ligand, antigen-antibody, DNA-protein, sugar-lectin, RNA-ribosome, etc. An example of molecular recognition is the antibiotic vancomycin that selectively binds with the peptides with terminal D-alanyl-D-alanine in bacterial cells through five hydrogen bonds. The vancomycin is lethal to the bacteria since once it has bound to these particular peptides they are unable to be used to construct cell wall. 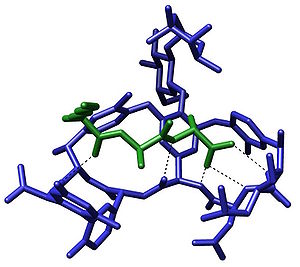
Another, out of countless possible examples: Zinc finger proteins coordinate one or more zinc ions to help stabilize their folds. The diagram represents the Cys2His2 zinc finger motif, consisting of an α-helix and an antiparallel β-sheet. The zinc ion (green) is coordinated by two histidine residues and two cysteine residues. 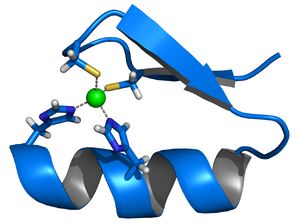
Zinc finger proteins typically bind DNA, RNA, proteins or small molecules. The diagram represents the protein Zif268 (blue) containing three zinc fingers in complex with DNA (orange). The coordinating amino acid residues and zinc ions (green) are highlighted. 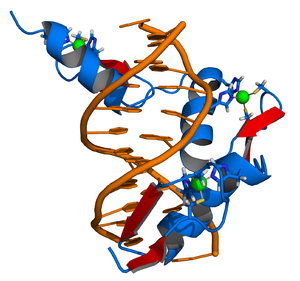 |
| Charge: | Complexes may be negatively charged, positively charged or they may be neutral. |
| Chemistry: | All biochemistry and molecular biology, totally outside the scope of this webbook! |
| Congeneric Series: | See above. |
Scaling Between Lewis Acid/Base Types
Lewis acid/base interactions can be scaled between general type/type interactions down to specific species/species interactions.
For example, a compound such as lithium chloride, LiCl, can be considered to be a Lewis acid/base complex, an s-LUMO/Lobe-HOMO Type 7 complex, a Group I cation/Group VIIA anion complex or as a Li+/Cl– complex.
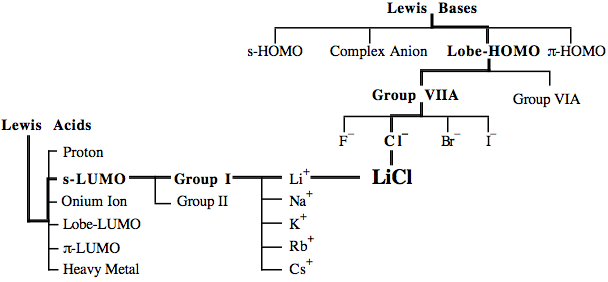
Hierarchical Families
Lewis acids, Lewis bases and Lewis acid/base complexes often exist in hierarchical families in which Lewis acid/base complexes are themselves chemical species able to behave as Lewis acids or bases and which therefore can be classified by type.
For example, the tetrahydroborate ion (or borohydride ion), [BH4]– is both a Type 13 complex and a complex anion Lewis base:
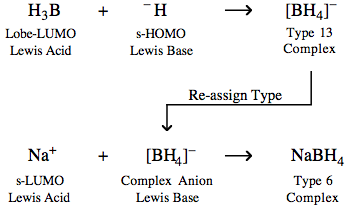
Multi-Step Reaction Mechanisms
Multi-step reaction mechanisms can be projected onto the matrix. Consider the alkylation of methoxybenzene (anisole) by 2-propylchloride and aluminium chloride. The reaction is a classic Lewis Acid catalysed electrophilic aromatic substitution reaction:
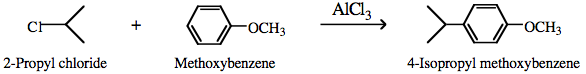
The alkylating agent, 2-propyl chloride, is a type 15 complex in which the carbon-chlorine bond is polarised.
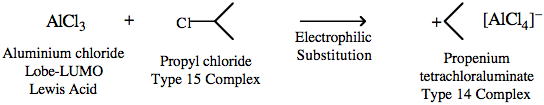
The first step of the reaction involves the Lobe-LUMO Lewis acid AlCl3 abstracting a chloride ion Lobe-HOMO Lewis base from propyl chloride to form a Type 14 complex. The driving force for this reaction is the formation of the stable [AlCl4]– ion.
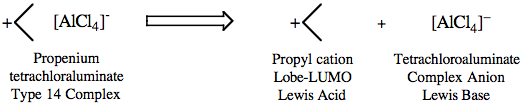
The type 14 complex can be deconstructed into its constituent parts: an electrophilic Lobe-LUMO (propenium) carbenium ion Lewis acid with a non-nucleophilic tetrachloroaluminate complex anion counter ion.
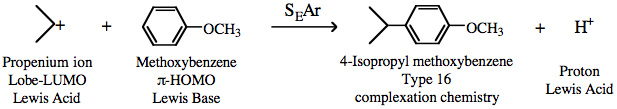
The propenium ion Lobe-LUMO Lewis acid then initiates an electrophilic substitution which begins as a lobe Lewis acid/π-Lewis base interaction, ie type 16 complexation chemistry:
PATTERNS IN REACTION CHEMISTRYThe central part of the chemogenesis analysis – the identification of the five reaction chemistries, the classification of Lewis acids and Lewis bases and the formation of the Lewis acid/base interaction matrix – has been published as a poster + book available from meta-synthesis. 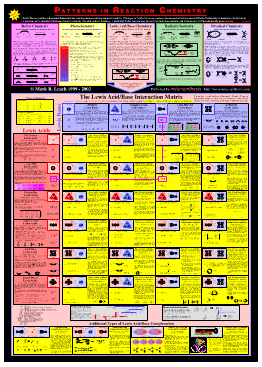 Buy the Lewis Acid/Base Reaction Chemistry Package (book + poster) The Lewis Acid/Base Reaction Chemistry Package is made up of two parts:

Need more information? Contact sales@meta-synthesis.com |

|
|
| Lewis Acids & Lewis Bases | Matrix Poster |
© Mark R. Leach 1999 –
Queries, Suggestions, Bugs, Errors, Typos...
If you have any:
Queries
Comments
Suggestions
Suggestions for links
Bug, typo or grammatical error reports about this page,please contact Mark R. Leach, the author, using mark@meta-synthesis.com
This free, open access web book is an ongoing project and your input is appreciated.