Periodic Table |
 |
 |
 |
 |
 |
 |
| The STAD Mechanistic Step | The Mechanism Matrix |
Unit & Compound Mechanistic Steps
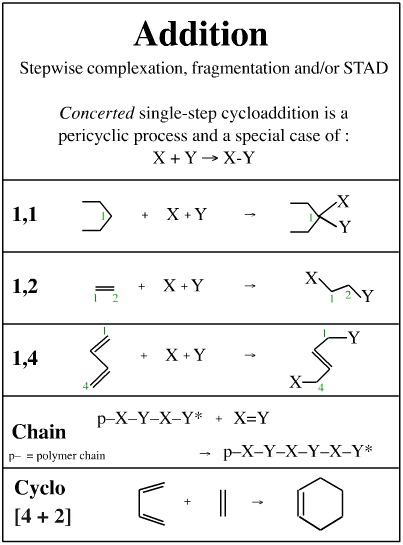
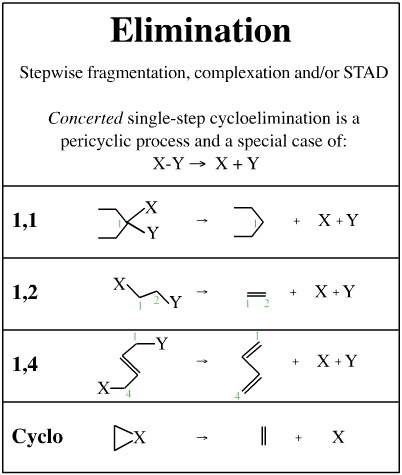
All mechanistic process can be deconstructed into sequences of substitution- transfer- abstraction- displacement or STAD steps, as suggested previously, however it remains convenient to classify mechanisms in terms of complexation, elimination, rearrangement, etc.Atom-to-Atom Mapping
Rigorous deconstruction to the STAD level is not particularly useful when trying to understand reaction mechanism science. It is better to consider mechanisms in terms of concerted unit mechanisms, and to construct multi-step compound mechanisms from sequences of these concerted unit mechanisms. We will examine:
Complexation
Fragmentation
Substitution
Insertion
Pericyclic processes
Metathesis
Addition
Elimination
Rearrangement
Name Reactions
It is not that aim of this page say everything-that-can-be-said about reaction mechanisms, that would be impossible. Instead, the emphasis will be on the various types of concerted and stepwise atom-to-atom mappings which are available, for example:
- Concerted vs. Stepwise substitution.
- 1,2-Addition vs, 1,4-addition, in preference to looking at the various subtypes of 1,2-addition.
- Single-step 1,2-addition vs. chain 1,2-addition.
- Fragmentation vs. complexation, and elimination vs. addition.
Little is said on this page about nucleophilic substitution or radical chain addition, etc., although it will sometimes be necessary to illustrate a general point with a specific instance.
Complexation
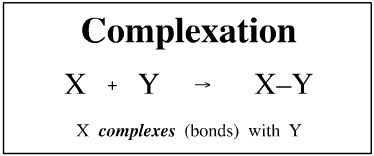
The complexation of Lewis acids and Lewis bases and radical coupling has been discussed at length elsewhere in this web book, here and here, and nothing will be added here about the types of complexation. In this section we are looking at X + Y → X-Y, pure and simple.
The first thing to say is that complexation is a fundamental, concerted, single-step, unit mechanistic process.
Complexation – by definition – involves bond formation, and bond formation is an exothermic process. The result that a complexation reaction will have a negative ΔH (enthalpy) value.
This must be the case because the bonded state complex, X-Y, will always have a lower energy than the free species X + Y, or the bonded state would not form. The law of conservation of energy tells us that this bonding energy must be released in some way, usually as heat.
But, X + Y involves two species, while X-Y involves only one. The result is that X + Y has more entropy – in other words the system is more dispersed – than the complexed form, X-Y.
The Gibbs free energy relationship, ΔG = ΔH – T DS, for this system tells us that complexation vs. decomplexation is a subtle balance between two components:
- Enthalpy, or heat of formation.
- Entropy, the tendency of the system to disperse.
- The crucial point is that the entropy component is multiplied by the thermodynamic temperature, so as the temperature increases the entropy component becomes more important and fragmentation dominates. Conversely, at low temperatures complexation dominates.
However, there is a third factor which is not explicitly dealt with by the Gibbs equation: activation energy. At room temperature, X and Y may coexist together indefinitely, while at 120°C the reaction to X-Y may proceed.
So, the X + Y → X-Y reaction may require moderate heating to overcome the activation energy of the reaction, but if the X–Y complex is heated too strongly it will invariably fragment to X + Y.
The enthalpy (heat of reaction) of the X + Y→ X-Y reaction must be removed in some way, or X-Y will fragment back to X + Y. This is not a problem in the liquid phase where the heat or formation can be conducted away by the solvent, but this is not the case in the gas phase the newly formed X–Y, or rather [X-Y]*, will be so vibrationally excited, or "hot", that it will fragment. The newly formed [X-Y]* must transfer this excess energy away to another species or the walls of the vessel. For this reason gas phase complexation reactions are often shown with a third species, m:
X + Y + m → X–Y + m*
In the reaction, m is heated to the species m* which carries away the excess kinetic and thermal energy, here. Although often shown as a three-centre process, the reaction is more likely to be stepwise, it all depends of the timing:
X + Y → [X–Y]*
[X–Y]* + m → X-Y + m*
Indeed, complexation only ever involves two species coming together single step (concerted) because the three centre complexation process:
X + Y + Z → X-Y-Z
is statistically unlikely.
Fragmentation

When an X + Y ⇄ X-Y reaction is at thermal equilibrium, fragmentation is the reverse of complexation. But this condition does not always hold. Fragmentation need not be the reverse of complexation because a chemical compound can fragment into any number of pieces.
Trinitrotoluene, TNT, C7H5N3O6, has a molecular weight of 227.13. On heating, the TNT molecule rapidly disassembles into a zoo of small, hot gas molecules: CO2, N2, NO2, H2O, etc.
The explosive fragmentation of TNT to thermodynamically stable molecules like N2 and CO2 is favourable on both enthalpy and entropy grounds.
Substitution

Substitution or ligand exchange reaction can either proceed via a concerted STAD mechanism or by a sequence of fragmentation-followed-by-complexation steps.
Consider a tetrahedral (sp3) carbon centre with four different ligands, L1, L2, L3 and X, arranged so the carbon centre is chiral and optically active (ie., a single enantiomer).
A reaction occurs where X is replaced by Y. This can occur in two general ways:
Y may complex with the carbon centre to give a hypervalent three-centre transition state that immediately collapses back to a tetrahedral configuration with the ejection of X. The rate of this concerted bimolecular reaction is dependent upon the concentrations of both [L3CX] and [Y] and so is classed as a Second Order substitution. The backside attack mechanism means that the carbon centre "inverts" to give a Y substituted product of opposite configuration and the carbon centre remains as a single, optically active enantiomer:
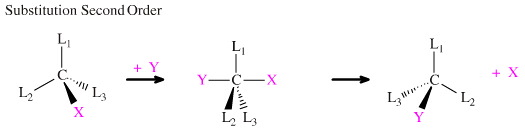
Note that the phrase "opposite configuration" is sensitive to the way in which configuration is defined, and there are three different systems: D/L, +/– or R/S. (The forth d/l system is the same as the +/– system.)
Alternatively, the substitution reaction may proceed in a stepwise manner with X leaving as a first step to give a hypovalent three-centre transition state, L3C, which subsequently complexes with Y. The rate of the reaction is dependent only upon the rate of fragmentation of [L3CX] and so is classed as a unimolecular or First Order process. The three centre intermediate can be attacked by Y from "either side" and this leads to the scrambling of the chiral centre to give a non-optically active, racemic, product mixture.
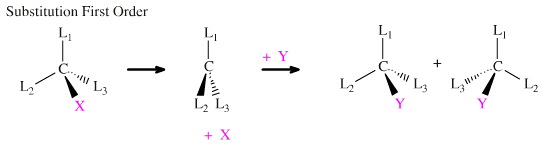
It is usually the case that:
• Concerted reactions are stereospecific and chirality retained.
• Stepwise reactions tend to racemise chiral centres which removes optical activity.
Ligand substitution reactions are also seen in inorganic octahedral complexes. Again, there are two possibilities:
Associative Interchange, Ia, which involves a hypervalent seven centred intermediate.
Dissociative Interchange, Id, which has a hypovalent five centred intermediate.
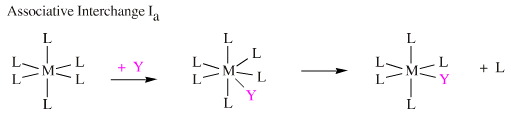
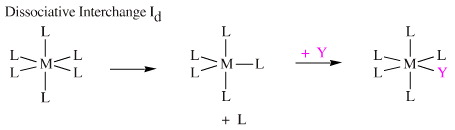
However, the difference between the Ia and the Id mechanisms is not nearly as clear cut as between first and second order substitution at a carbon centre, and there are many intermediate cases. Inorganic ligand substitution mechanisms are discussed in considerable detail, here.
Ligand interchange also occurs at square planar complexes:
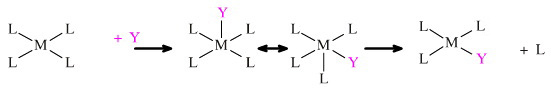
Square planar complexes are commonly associated with Ni, Pd, Pt (and other) centres.
Ligands substitute in a stepwise or one-at-a-time manner, and the intermediate compounds can be isolated and purified. It has been found that the second substituting ligand has a choice and can substitute in two ways so that the disubstituted square planar complex can have either a cis or a trans configuration.
Furthermore, it has also been found that when going from MX4 → MY4 the disubstituted intermediate may be trans, but when going from MX4→ MY4 the disubstituted intermediate may be cis.
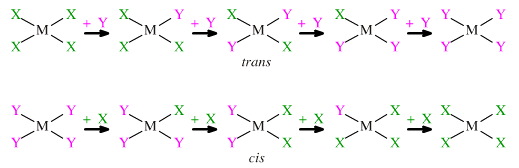
The "trans effect" seen with chloride and ammonia ligands displacing each other at a platinum(II) centre. And this chemistry is not simply of dry academic interest because the cis platinum complex, cis diamminedichloroplatinum (or DDP):
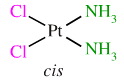
is an important and widely used anticancer drug known as cisplatin, search here.
Insertion

When a chemical species "squeezes-in" between two existing species, the reaction can be classed as an insertion. However, true, concerted reactions are comparatively rare and most examples are seen in organometallic chemistry and with diradicals.
• If the insertion proceeds as a single step, then the insertion process can be regarded as a special case of complexation, and the temperature dependence arguments associated with complexation apply, here.
• Insertion can also be regarded as 1,1-addition.
• Insertion can also be regarded as a redox process because it is usual for the oxidation and/or coordination number of the inserted species to increase.
• Insertion can occur via a sequence of mechanistic steps with insertion only as the overall result.
The direct insertion of a species into a chemical bond is common in organometallic chemistry where metals frequently insert into carbon halogen bonds. The process is called metallation.
Magnesium metal, Mg, inserts into the carbon iodine bond of methyl iodide, H3C-I, to give the well-known Grignard reagent, methyl magnesium iodide. This process can also be classed as an oxidative addition because the oxidation state of the magnesium increases from 0 to +2. The reaction is thought to involve single electron transfer (SET).
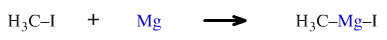
In recent years there has much interest in organopalladium chemistry. The first step of the synthetically useful Heck reaction involves a palladium(II) salt inserting into an aromatic halogen bond. This insertion (which has been simplified in the diagram below) is also classed as an oxidative addition because the palladium is oxidised from +2 to +4.
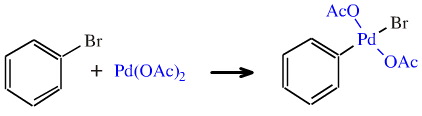
Consider the reaction of phosphorus trichloride, PCl3, with chlorine, Cl2, to give phosphorus pentachloride, PCl5. In this reaction, which is often used to illustrate chemical equilibrium, the PCl3 [can be considered to] insert into the Cl-Cl bond, with the oxidation number of the phosphorus increasing from +3 to +5.
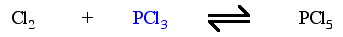
Carbenes are very reactive diradicals species, and it has been suggested that the parent carbene species, methylene or CH2, is the most reactive of all organic moieties. Methylene – which is divalent – can insert into alkane C–H and C–C bonds to give a tetravalent species. However, these reactions are very unselective and of little synthetic utility:
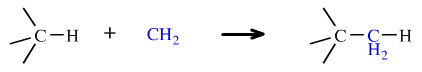
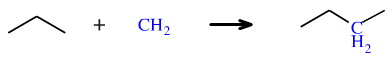
Dichlorcarbene, CCl2, is less reactive than methylene. It is generated from chloroform (trichloromethane, CHCl3) and strong base which initiates the 1,1-elimination of HCl:
CHCl3 + NaOH → CCl2 + NaCl + H2O
Dichlorocarbene is able to react with the heterocycle pyrrole to give 3-chloropyridine, admittedly in low yield:
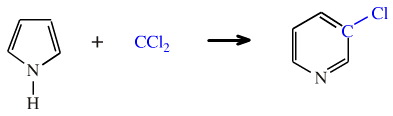
This Reimer-Tiemann reaction is, overall, an insertion of carbon into the aromatic ring system which expands from a 5 to a 6 membered system. However, the reaction proceeds by quite a number of discrete steps.
Another example of multistep insertion is the oxidation of an "allylic" carbon to a hydroperoxide, here:
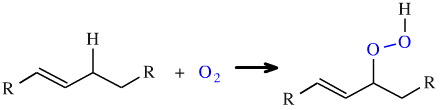
This oxidation process is very important in the food industry where it is involved in the degradation of unsaturated fats and oils.
Pericyclic Processes

Pericyclic reactions have a cyclic transition state which allows for the concerted rearrangement of the electrons so that sigma and π-bonds to simultaneously break and form. Four subclasses of pericyclic reaction can be recognised and we will use this system:
- Cycloaddition
- Electrocyclic Reactions
- Sigmatropic Rearrangements
- Group Transfer Reactions
Pericyclic reactions are discussed in their own section, here.
Metathesis Reactions

The classic metathesis reaction is also called a double displacement reaction. This type of metathesis occurs when aqueous solutions of two soluble salts are added together and an insoluble precipitate appears:
When aqueous solutions of lead(II) nitrate, Pb(NO3)2, and potassium iodide, KI, are mixed, a precipitate of lead(II) iodide, PbI2, appears and the potassium nitrate, KNO3, remains in solution. The ions "swap". Likewise, mixing aqueous solutions of silver nitrate, AgNO3, and sodium chloride, NaCl, gives a precipitate of silver chloride, AgCl, and sodium nitrate, KNO3, remains in solution:
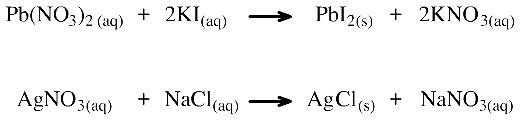
The driving force for these reactions is the formation of a low solubility precipitate. If there where to be no precipitate the four species (two cations and two anions) would simply remain in solution.
Selective precipitation reactions can be used to identify metal ions in solution, and extensive analytical methodologies have been developed using this approach, see Qualitative Inorganic Analysis by Vogel). However, these wet chemistry techniques have fallen out of use because instrumental methods of analysis such as atomic absorption spectroscopy and ICP-MS, are: easier to perform, faster, more accurate, more precise, and they are far, far more sensitive.
Substituted alkenes (olefins) undergo olefin metathesis, a reaction catalysed by transition metals (often Mo, W or Re) in the presence of a trialkyl aluminium, AlR3, alkylating agent. During the metathesis the alkene ligands are exchanged between the alkenes. The reaction process is most clearly visualised when a cyclic alkene undergoes olefin metathesis with a linear alkene. The product is a linear diene, but there are many by-products.
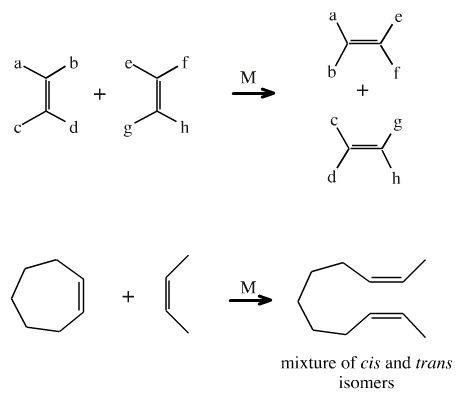
Note that in the diagram above, the alkene double bonds are shown cis, but this is only for clarity. Mixtures are cis and trans isomers are formed.
It is tempting to postulate a cyclic four membered "pericyclic type" transition state associated with the transition metal centre... but kinetic measurements and examination of product ratios indicates that this is not the case. It is more likely that the reaction is stepwise, and involves metal=carbene intermediates.
Yves Chauvin, Robert H. Grubbs, and Richard R. Schrock shared the 2005 Nobel Prize in Chemistry for "the development of the metathesis method in organic synthesis". Wikipedia
Addition Reactions
Addition reactions usually involve two species complexing to the two ends of a π-system, with the π-system correspondingly reducing in length. Typical reactions of this type would be the addition of bromine, Br2, to a 1,4-diene:
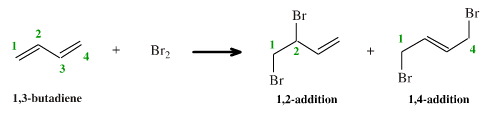
There are two products, one formed by 1,2-addition and the other formed by 1,4-addition.
In each case the π-systems reduces from a 4 π-electron system to a 2 π-electron system.
The numbers "1,2" and "1,4" refer to the π-system and should not be confused with the numbering system used to name the overall molecule.
Using the addition reaction nomenclature system, we can consider the reaction of carbon monoxide, CO, with chlorine to give phosgene to be a 1,1-addition:
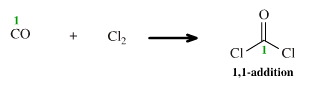
However, this reaction can also be considered to be an insertion of carbon monoxide into the Cl-Cl bond, or as a special case of complexation.
Addition reactions are net bonding processes and are thermodynamically similar to complexation, here.
Play with the Addition Reaction Synthlet.
Cyclohexene: A Model System
Cyclohexene is a wonderful model system to use for the study the subtleties of addition. At the simplest level, a species X2 adds to cyclohexene to give a 1,2-disubstituted cyclohexene.
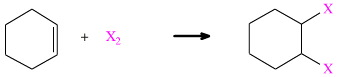
There are several examples: Cl2, Br2, I2 and H2 all add to cyclohexene in this manner. Chlorine adds to give 1,2-dichlorocyclohexane. Hydrogen adds – with the help of a Pt catalyst – to give cyclohexane.
It is plausible for X2 to add to cyclohexene in two ways: syn and anti.
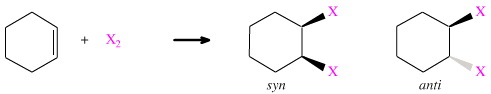
syn-Addition is deemed to occur when the two X atoms add to the same face of the cyclohexene ring, the result is a cis-1,2-disubstituted cyclohexane.
anti-Addition is deemed to occur when the two X atoms add to opposite sides of the cyclohexene to give a trans-1,2-disubstituted cyclohexane.
There is an important steric constraint if the two X functions to add in a syn manner: they must be connected in some way. Either the two X functions must be part of the same molecule, in which case the reaction is a cycloaddition, or they must be served in some syn manner. An example would be catalytic hydrogenation where hydrogen is first adsorbed onto a metal surface, from where it is received by the alkene in a syn manner.
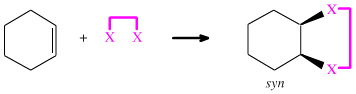
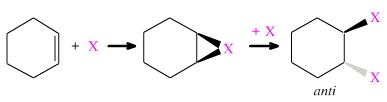
Conversely, to form the anti-addition product, the two X functions cannot be connected and the reaction must proceed in some kind of stepwise manner:

Stereochemical control of stepwise anti-addition can be maintained by first applying concerted syn cycloaddition, and then ring opening via concerted substitution:
Consider a polar species, X-Y, adding to 1-methyl cyclohexene, where X is electronegative and Y is electropositive. Examples of polar-species-which-add would include: H-Cl, H-Br or H-OH (H2O). There are two possible modes of addition:
- Markovnikov addition where the electronegative atom, X, adds to the more substituted carbon centre.
- Anti-Markovnikov addition where the electronegative atom, X, adds to the less substituted carbon centre.
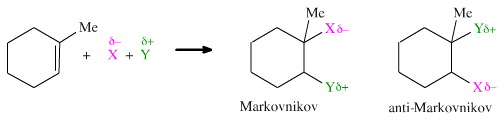
The syn and anti type additions can be combined with the Markovnikov and anti-Markovnikov type additions to give a number of possible products. Consider the 1-methylcyclohexene with an additional bulky group, R, to make the molecule chiral (which actually makes the analysis easier.) Consider the addition of water. There are six possible H2O addition products for this system:
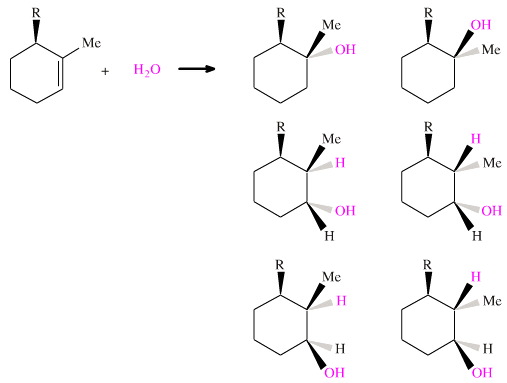
Using a variety of reagents and techniques, it is possible make all six of the alcohols stereospecifically. But they cannot not all be made in a single preparative step.
Chain Addition
1,2-Addition reactions can chain propagate to produce polymers:
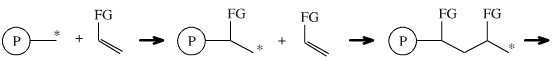
A growing polymer chain with a reactive centre, *, adds to an alkene, or a functionilised alkene, where FG can be Cl, CN, C6H5, etc., to produce a chain two carbon atoms longer with a new reactive centre which is able to partake in another addition... and so on...
Depending upon the nature of the FG and the initiator, 1,2-chain addition can be cationic, radical or anionic in nature, and examples of each type are known:
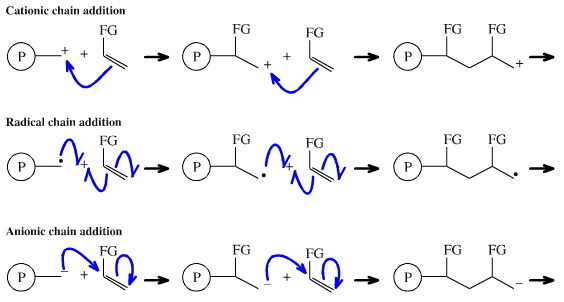
Concerted, single step cycloaddition reactions are pericyclic processes (see above).
Addition proceeds via single or sequential complexation, and is an exothermic process:
Like complexation, an elevated temperature may be needed to overcome the reaction's activation energy, but very high temperatures will cause elimination.
Elimination Reactions
It is tempting to think of elimination as the opposite or reverse of addition because 1,2-addition and 1,2-elimination reactions can be complementary. For example, ethene, CH2=CH2, reacts with hydrogen chloride, HCl, to give chloroethane... and chloroethane is able to lose HCl to give ethene:
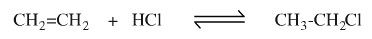
But the ionic mechanisms are entirely different:
The forward reactions proceeds by two discrete steps: protonation of the electron rich π-system to give the carbenium ion intermediate which is subsequently complexed by the chloride ion:
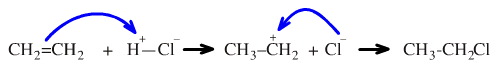
The reverse reaction proceeds rather easily through base catalysed beta-elimination. This concerted reaction involves a base abstracting a beta-proton. In one concerted step the proton is removed, the π-bond is formed and the chloride ion is ejected:
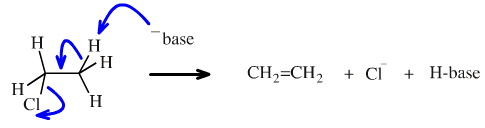
The concerted beta-elimination mechanistic process cannot happen in reverse (in a homogeneous environment), because the proton, chloride ion and alkene would have to find themselves precisely aligned in space, statistically a very unlikely scenario.
Elimination is a type of fragmentation and by whatever mechanism it occurs (beta-elimination, retrocycloaddition or stepwise elimination) is encouraged on thermodynamic grounds by increasing temperature and decreasing pressure:
The products of elimination have more entropy.
Rearrangement Reactions
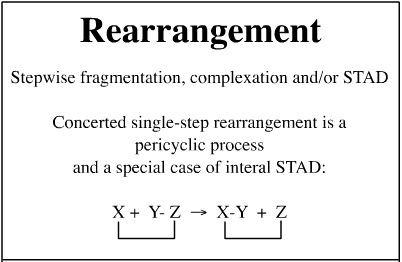
A rearrangement is deemed to have occurred when a function, R, moves along a chain of atoms.
[1,2] Rearrangements
Schematically, the [1,2] rearrangement involves R moving to an adjacent atom:
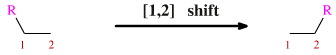
There quite a number of named [1,2] rearrangements which occur to specific organic functional groups under specific conditions. Some of these name reactions proceed in a concerted manner via cyclic intermediates and some may be stepwise:
Wagner-Meewein
Hofmann
Curtis
Schmidt
Beckmann
Darkin
Lossen
Baeyer-Villiger
Stevens
Wittig
Favorskii
Pinacol-Pinacalone
Overall, these can be represented by:
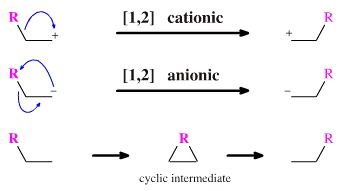
[1,j] Rearrangements
[1,j] Rearrangements occur when a function, R, moves along a chain of j atoms.
While there is great variety amongst [1,2] shift rearrangements, the longer migrations invariably proceed via a concerted pericyclic mechanism, here, however, the charges and bonds can be accounted for using curly arrows:
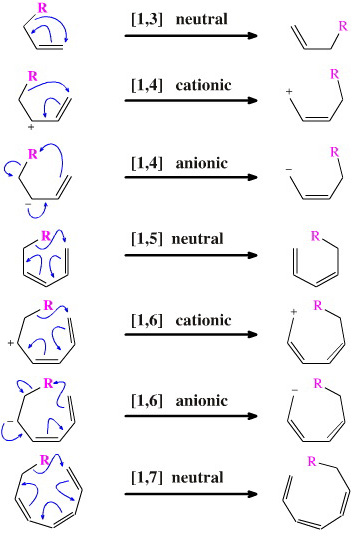
There are some interesting reactivity patterns with [1,j] rearrangements:
- [1,2], [1,4] and [1,6]
rearrangement requires a cationic or anionic centre.
- [1,3], [1,5] and [1,7]
rearrangements can occur with neutral systems. (Note
that with these neutral rearrangement systems, the curly arrows can
be constructed in equivalent clockwise or anticlockwise directions,
but only one is shown.)
- These pericyclic reactions can be activated by heat or UV light. The Woodward-Hoffmann rules can be used to predict the stereochemical consequences, see here
It is interesting to ask why a rearrangement reaction should proceed at all, because the way the reactions diagrams are drawn in the diagram above, there is no apparent energy difference between the left and side and the right hand side of the reaction equations, and so no driving force.
Consider the reaction of butane with aluminium chloride plus a catalytic quantity of 1-chlorobutane at 150°C. These conditions give an equilibrium mixture consisting of 20% butane (n-butane) and 80% methylpropane (iso-butane) .
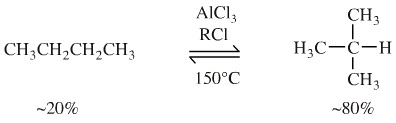
The reaction, a Wagner-Meewein rearrangement. It proceeds by the aluminium chloride abstracting a chlorine anion from the RCl to generate a carbenium ion. The carbenium ion abstracts a hydride from butane to produce a [C4H9]+ carbenium ion. This carbenium ion can [1,2] shift a methyl function to give (after hydride abstraction) the branched isobutane:
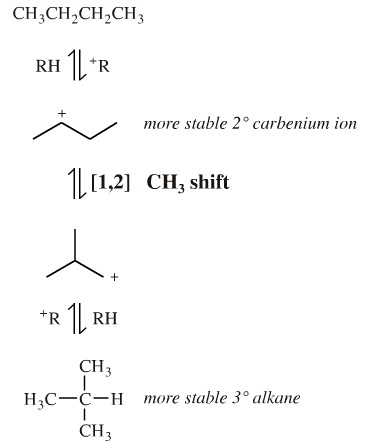
This apparently simple example of a [1,2] shift is actually quite involved because the product ratio is the balance of two competing stability issues:
- Branched alkanes,
with tertiary and quaternary carbon centres, are thermodynamically
more stable than linear alkanes. Butane has an energetic tendency
to isomerise to methylpropane.
- Secondary carbenium ions, R2HC+, are more stable than primary carbenium ions, RH2C+.
R3C+ > R2HC+ > RH2C+ > H3C+
In isolation the equilibrium position of the [1,2] rearrangement lies to the side of linear butane 2° carbenium ion, but the equilibrium is pulled over to methylpropane due to the greater thermodynamic stability associated with the more substituted carbon centre.
Note. This reaction tells us that methyl groups must have a much greater tendency to migrate than hydrogen under carbenium ion conditions. If this were not so the the most stable carbenium ion, the tertiary butyl cation, (CH3)3C+, would form leading to 100% methylpropane (isobutane).
The thermodynamic stability of branched alkanes over linear alkanes is exploited in the petrochemical industry where the process of hydrocracking is used to convert longer, linear alkanes into shorter branched alkanes. A wide range of materials can be produced and the process can be optimised for gasoline or kerosene production. Hydrogen, shape selective zeolite catalysts, 340-450°C cracking temperatures and 80-200 bar pressures are used.
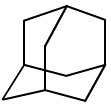
| Adamantane Wikipedia
There is an amusing synthetic chemistry story that illustrates how the subtle kinetic and thermodynamic effects associated with rearrangement reactions can be exploited. Synthetic chemists enjoy making target molecules, usually natural products or molecules with interesting symmetry. One such target was adamantane, C10H16, a beautifully symmetric hydrocarbon which is the hydrocarbon derivative of a single unit cell of diamond:
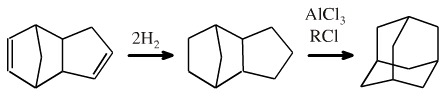
A multistep synthesis of adamantane was successfully devised, implemented and the results published. Success! Only... a few years later it was discovered that if the dimer of cyclopentadiene (discussed here) is hydrogenated and treated with AlCl3 + RCl, a series of Wagner-Meewein cationic [1,2] shifts occur and adamantane is produced in 15% yield:
It was subsequently demonstrated that any C10H16 alkane will rearrange to a 15% equilibrium mixture of adamantane under Wagner-Meewein conditions. |
[i,j] Rearrangements
Rearrangements can occur in which two chains "move with respect to each other"... although this is easier to explain and visualise with graphics than words:
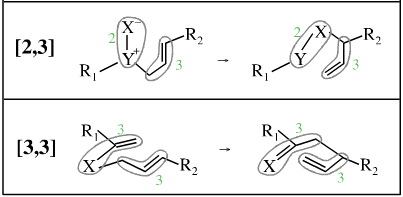
In these rearrangements a "two" unit moves with respect to a "three" unit hence [2,3], and a "three" unit moves with respect to a "three" unit, hence [3,3].
[3,4], [3,5], [4,5], [5,5], [9,9] etc. rearrangements are all known. All of these should be considered as sigmatropic processes, which are discussed here.
Name Reactions

Many, many many reactions only occur to specific functional groups under specific conditions and they usually involve quite a number of discrete mechanistic steps.
It is common to refer to such reactions by a name... often the name of the chemist who first observed or exploited the reaction.
There are many websites which deal with organic name reactions:
- Wikipedia List of Organic Reactions
- Organic Chemistry Portal
- Monomer Chem's Name Reaction List
- NameReaction.Com
- etc
|
|
| The STAD Mechanistic Step | The Mechanism Matrix |
© Mark R. Leach 1999 –
Queries, Suggestions, Bugs, Errors, Typos...
If you have any:
Queries
Comments
Suggestions
Suggestions for links
Bug, typo or grammatical error reports about this page,please contact Mark R. Leach, the author, using mark@meta-synthesis.com
This free, open access web book is an ongoing project and your input is appreciated.