Periodic Table |
 |
 |
 |
 |
 |
 |
| Addition Synthlet | Why Reactions Occur |
Pericyclic Reaction Chemistry
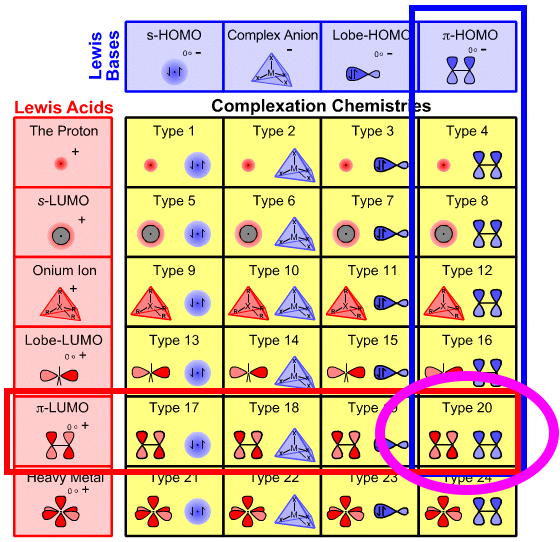
Pericyclic reactions represent an important class of concerted (single step) processes involving π-systems. The fact that the reactions are concerted gives fine stereochemical control of the product, however, this page is more concerned with the general types of pericyclic reaction, than with regio and stereochemical control. Many types of pericyclic reaction can be considered in terms of Type 20 [π-LUMO Lewis acid /π-HOMO Lewis base] interactions:
Introduction
- Pericyclic reactions have a cyclic transition state.
- While in this transition state, a concerted rearrangement
of the electrons takes place that causes σ and π-bonds
to simultaneously break and form.
- Pericyclic reactivity can be understood in terms of frontier molecular orbital (FMO) theory and the outcome of reactions can be predicted using the Woodward-Hoffmann rules.
Pericyclic reactions are popular with synthetic chemists because the reagents and conditions are mild and the reactions are usually very "clean"... unlike so many organic chemical reactions that result in the formation of large quantities of brown-black, smelly by-product of unknown composition...
In Frontier Orbitals and Organic Chemical Reactions, J.Wiley & Sons (1976), Fleming recognises four subclasses of pericyclic reaction and we will use this classification system:
Cycloaddition
Electrocyclic Reactions
Sigmatropic Rearrangements
Group Transfer Reactions
Within each subclass, it is common that reactions can either be induced to occur under thermal conditions with simple heating, or under photochemical conditions. The two methodologies are complementary.
Cycloaddition
Cycloaddition, and the reverse process retrocycloaddion, can be observed in the reaction between 1,3-butadiene and ethene to give cyclohexene.
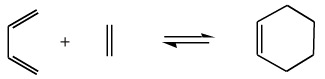
The 1,3-butadiene is a conjugated π-system with 4 π-electrons and the ethene is a conjugated π-system with 2 π-electrons. The reaction between 1,3-butadiene and ethene to give cyclohexene is described as a [4+2] cycloaddition reaction. This type of cycloaddition is also called a Diels-Alder reaction.
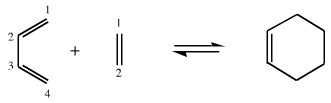
In a Diels-Alder reaction the 4 π-electron system is referred to as "the diene" and the 2 π-electron system as the "dieneophile". These terms are used in related [4+2] reaction systems even when the functional groups are not actually dienes or alkenes.
Cycloaddition is a type of X + Y → X-Y complexation, and it follows the usual thermochemistry rules.
The formation of the Diels-Alder adduct is an exothermic reaction.
It follows that high temperatures favour retrocycloaddition and low temperatures favour adduct formation.
However, many cycloaddition reactions require moderate heating to overcome the activation energy. So a cycloaddition may require heating to make the reaction "go", but if it is heated too much the equilibrium will favour retrocycloaddition.
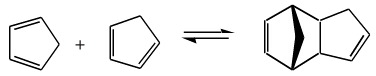
The compound cyclopentadiene slowly undergoes cycloaddition with itself: one molecule of cyclopentadiene acts as a 4 π-electron diene and the other as a 2 π-electron dieneophile. The product is a Diels-Alder "adduct", often called dicyclopentadiene. This dimeric material can be cracked back to cyclopentadiene by heating at 150°C for an hour and then distilling off the diene monomer.
Diels-Alder cycloaddition reactions proceed more efficiently if the diene is electron rich and the dienophile is electron poor. Cyclopentadiene is electron rich. The way to make the dieneophile electron poor is to add electron withdrawing groups, such as carbonyl functions.
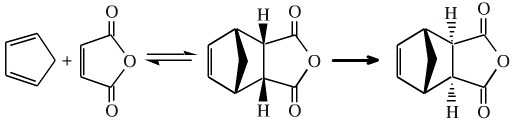
Maleic anhydride is an electron poor dieneophile which reacts with cyclopentadiene to give an endo Diels-Alder adduct. Upon heating at 190°C, the endo conformation adduct adopts the more stable exo-adduct conformation.
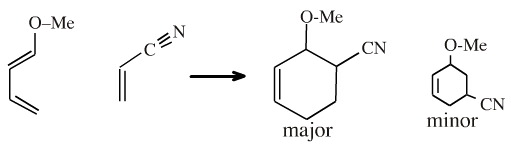
1-Methoxy-1,3-butadiene reacts with acrylonitrile to give 3-methoxy-4-cyanocyclohexene rather than the 3-methoxy-5-cyanocyclohexene isomer. This "ortho" regioselectivity of this reaction can be rationalised using FMO theory.
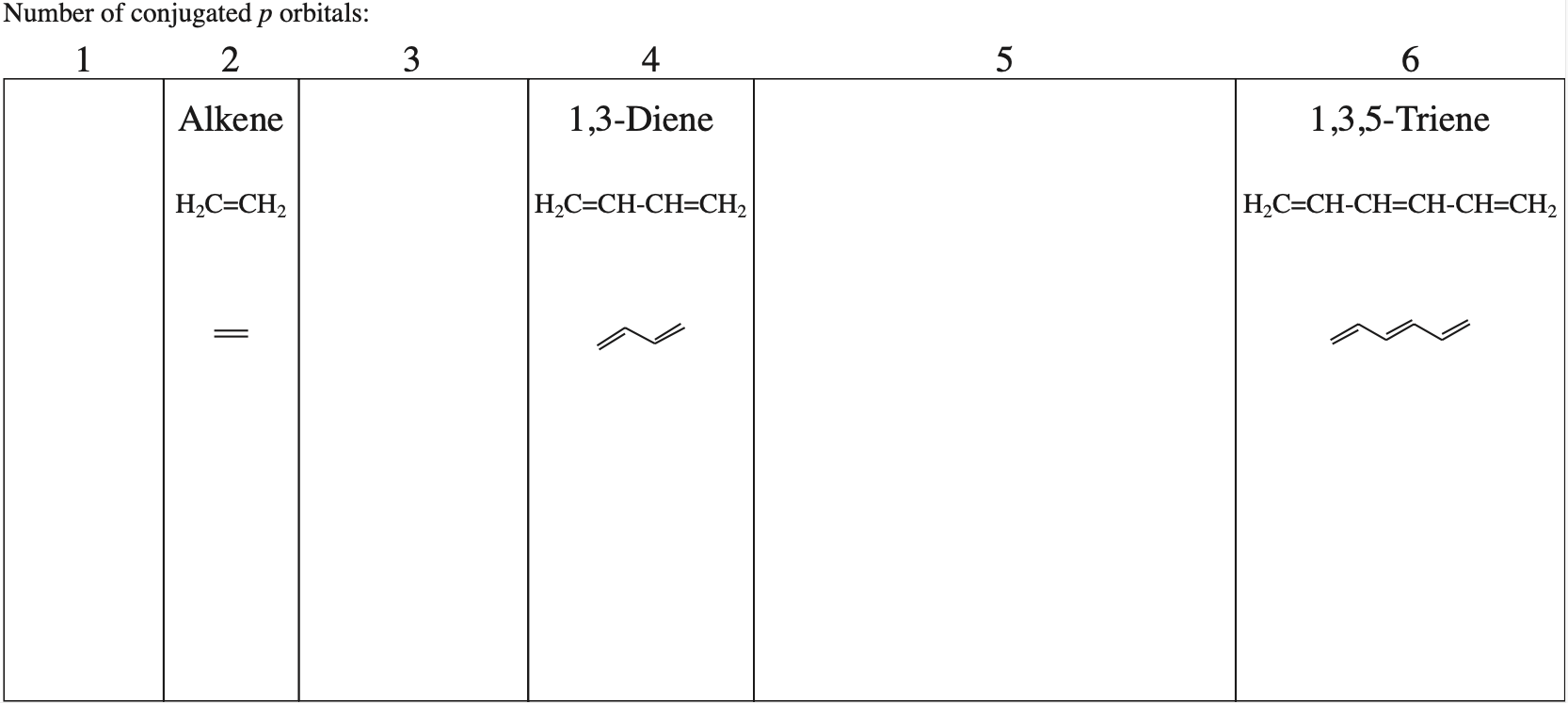
π–Molecular Orbitals
Consider the π–molecular orbitals of alkenes, dienes and trienes and the phases of the HOMOs & LUMOs:
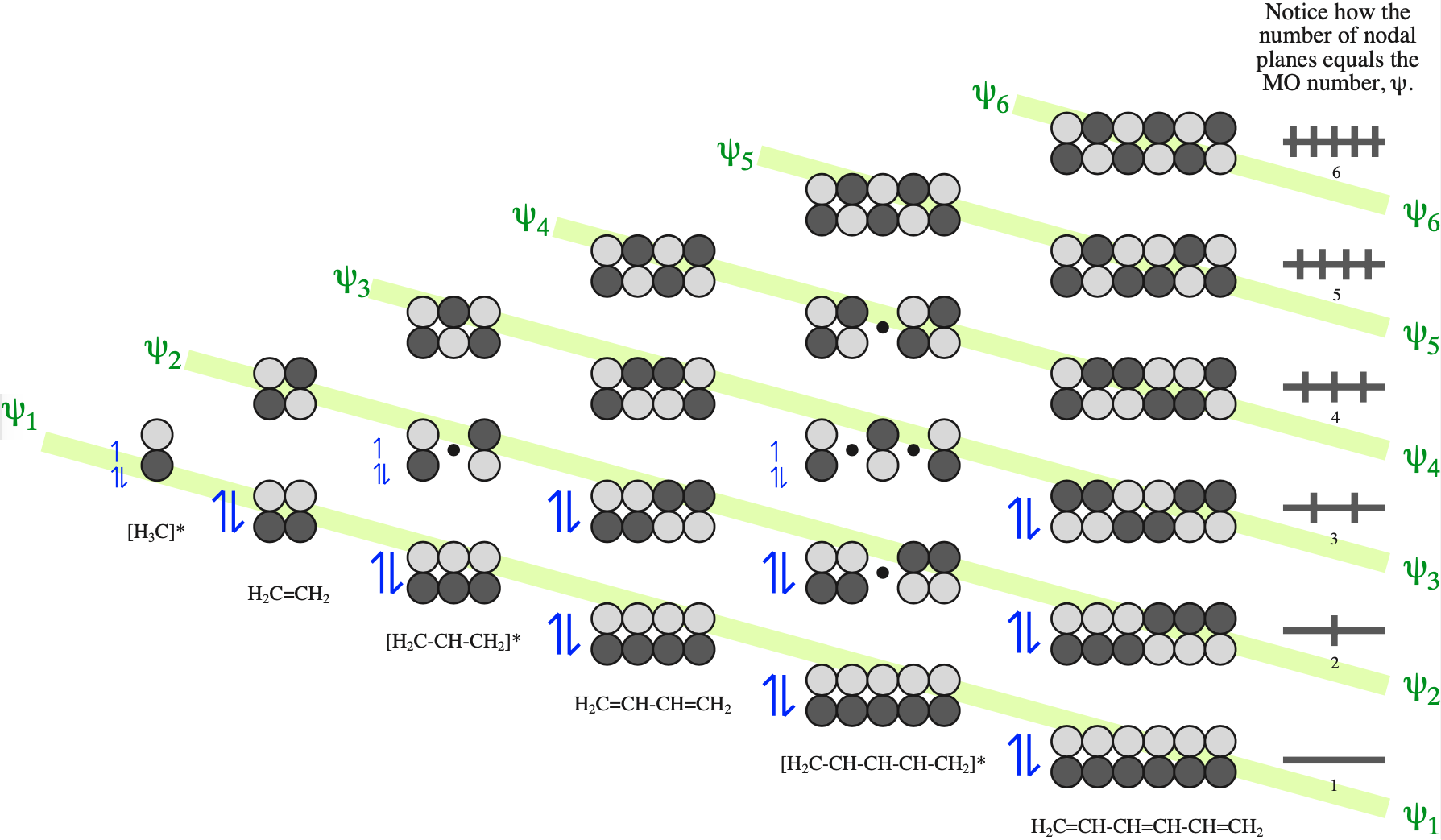
FMO Theory
Cycloaddition can be explained using frontier molecular orbital (FMO) theory.
|
Frontier Molecular Orbital Theory was developed in the 1960s by Kenichi Fukui who recognised that chemical reactivity can often be explained in terms of interacting Highest Occupied MOs (HOMOs), Lowest Unoccupied MOs (LUMOs) and Singly Occupied MOs (SOMOs).
The FMO approach was developed by Woodward & Hoffmann in the late nineteen sixties who used it to explain an apparently diverse set of reactions involving π-systems, including Diels-Alder cycloaddition. Hoffmann used the approach to explore transition metal complexes.
|
Using the Chemogeneis Lewis acid/base analysis logic, cycloaddion is a π-LUMO Lewis acid + π-HOMO Lewis baseType 20 Lewis acid/base complexation interaction:
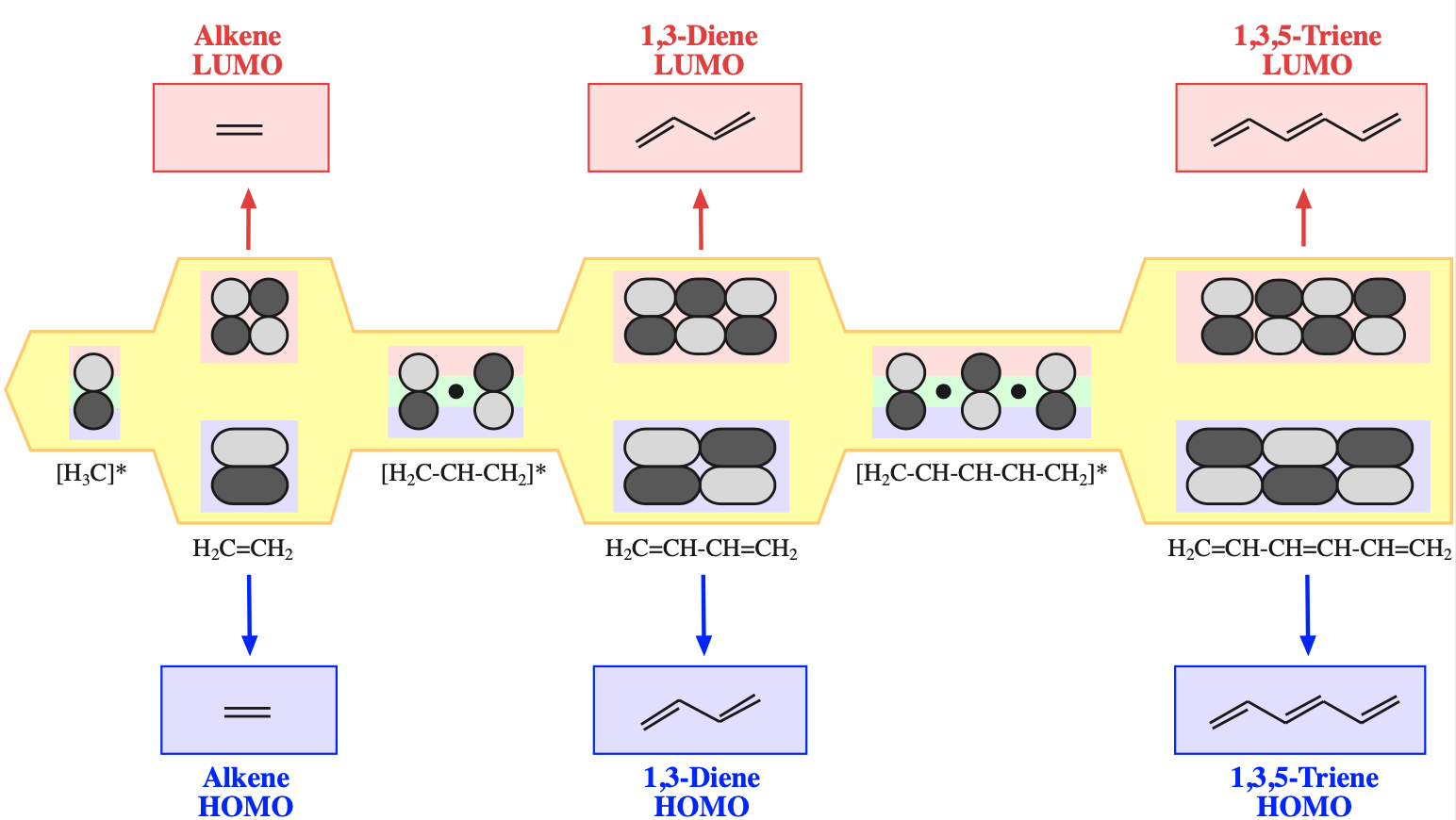
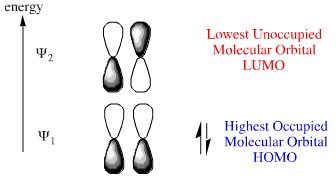
The alkene (dienophile) component has two electrons is a "single" π-bond. FMO theory, here, identifies the HOMO and LUMO components of this system:
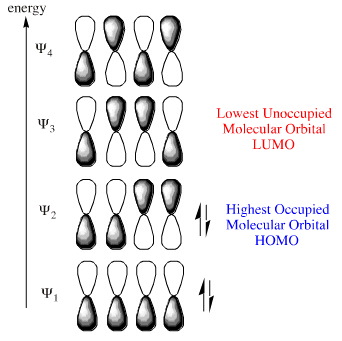
Likewise, the diene which has four electrons in is conjugated π-system can have its HOMO and LUMO identified within FMO theory:
If we examine the phases at the ends (termini) of the diene and dieneophile we find that the LUMO/HOMO interactions are phase matched:
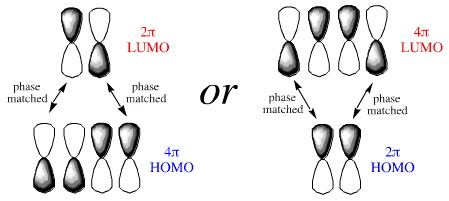
Notice that the phases match whichever species is defined as the HOMO or LUMO. In reality, the electron rich species reacts via its HOMO and the electron poor species via its LUMO.
In the FMO diagrams above, the sizes or coefficients, are all the same size, but usually they are of different sizes. The rule is that the coefficients match as well: small with small and large with large:
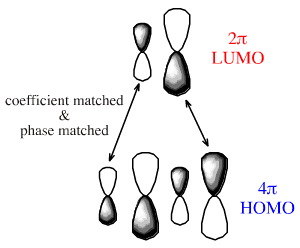
We can use Hückel MO theory to calculate the sizes of the coefficients at each of the atoms. (There is a web based HMO calculator, here, although at meta-synthesis we use a stand alone package: HMO by Allan Wissner.)
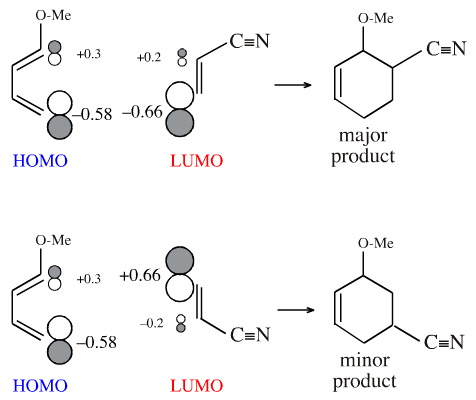
If the coefficients are calculated for 1-methoxy-1,3-butadiene the termini are +0.3 and –0.58 (or –0.3 and and +0.58). For acrylonitrile the coefficients are +0.2 and –0.66 (or –0.2 and +0.66).
The cycloaddition reaction proceeds so the coefficients "match", both in in terms of phase (essential) and in terms of coefficient magnitude: +0.3 with +0.2 and –0.58 with –0.66.
Thus, the regioselectivity of the cycloaddition can be explained:
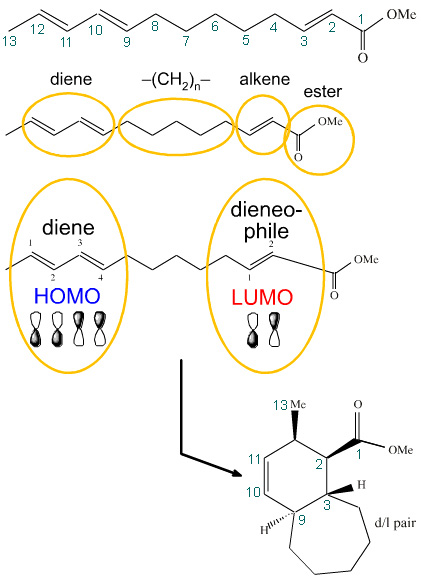
Cycloaddition is popular in the synthetic methodology when attempting to make natural products and pharmaceutical agents with involved stereochemistry because the reaction can determine the relative configuration of up to four chiral centres in a single reaction. Intramolecular cycloadditions can show particularity selectivity:
There are the HOMO and LUMO of this structure, calculated in Spartan:
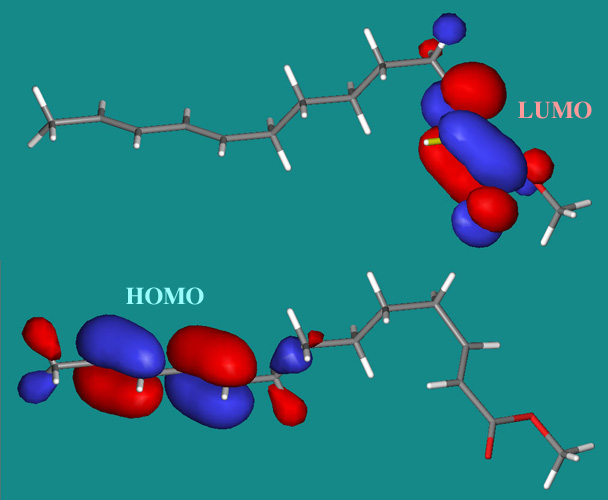
Cycloaddition reactions can be described in terms of π-LUMO/π-HOMO interactions. Thus they can be considered as Type 20 Lewis acid/base complexes, here.
A nice YouTube video from That Chemist:
Thermal and Photochemical Reactivity
Using the FMO logic discussed above, some π/π interactions are FMO symmetry forbidden and do not result in cycloaddition. Consider dimerisation of ethene to cyclobutane.
This reaction does not occur in either the forward or reverse directions under thermal conditions:
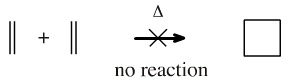
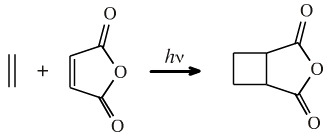
But... the [2+2] cycloaddition reaction does occur under photochemical conditions:

Ethene can [2+2] cycloadd to maleic anhydride to give the cyclobutane diacid anhydride (UV light, –65°C, 44 hours, 77% yield):
The FMO argument is that the LUMO and the HOMO of ethene are phase mismatched and cycloaddition is symmetry forbidden. But a photon promote one of the electrons into the LUMO making a transient diradical species. The excited state HOMO is now phase matched to combine with the LUMO and cycloaddition is symmetry allowed:
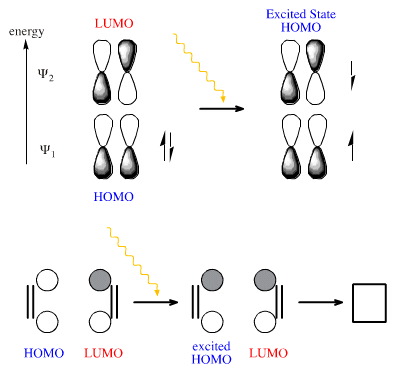
[2+2], [4+4] and [6+6] cycloadditions are thermally disallowed, but photochemical examples are known.
Electrocyclic Reactions
Electrocyclic reactions are unimolecular processes which involve the exchange of π-bonds for ring-closing sigma-bonds. This is best illustrated by an example:
Upon heating, 1,3,5-hexatriene will undergo an electrocyclic ring closure to give 1,3-cyclohexadiene:
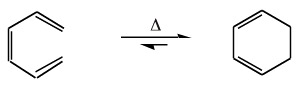
Note that the 3-alkene must be cis for the reaction to occur.
The reverse, or retroelectrocyclic, reaction can also occur. This is seen with the ring opening of cyclobutene to 1,3-butadiene:
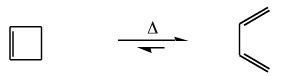
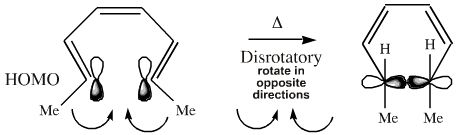
Electrocyclic reactions, like all pericyclic processes, exhibit great stereoselectivity. Consider two 1,3,5-hexatriene systems embedded into longer hydrocarbon chains.
trans-cis-trans-2,4,6-Octatriene will ring close to give a cis ring:
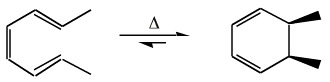
trans-cis-cis-2,4,6-Octatriene will ring close to give a trans ring:
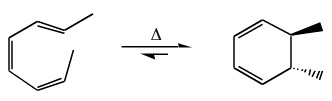
As with cycloaddition, this selectivity can be explained by examining the FMOs, specifically the HOMO:
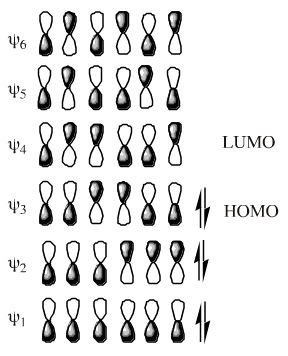
If the termini of the HOMO is superimposed upon the triene system, it can be seen that the end groups must rotate in a disrotatory manner (twist in opposite directions, when viewed front-on) to form the bond:
However, electrocyclic reactions can also occur photochemically. When photoactivated, an electron moves from the HOMO to the next orbital, the LUMO. (Now this orbital contains an electron it is no longer unoccupied, it is either a SOMO or an excited state HOMO).
The photoexcited system will ring close in the opposite manner to the thermal system and the groups conrotate (twist the same way) to form the sigma bond:
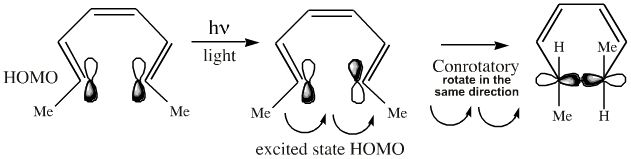
The two products have different stereochemistry and are diastereomers.
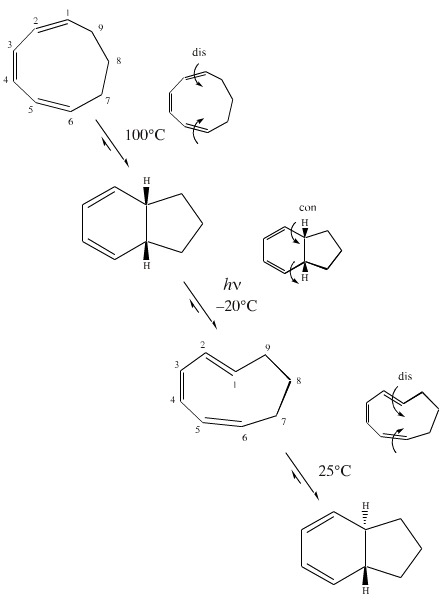
This thermal and photo selectivity can be exploited in the reaction sequence below in which 1,3,5-cyclononatriene is converted into bicyclic systems, first with cis and then trans ring junctions.
The initial 1,3,5-cyclononatriene is all cis. This is thermally ring closed in a disrotatory manner and then photo-ring opened at -20°C in a conrotatory manner. The 1,3,5-cyclononatriene now has two cis and one trans double bonds, however, the nine membered ring is able to accommodate the strain of a trans alkene. Thermal ring closure gives the trans-ring junction.
Sigmatropic Rearrangements
Like electrocyclic reactions, sigmatropic rearrangements are unimolecular processes. Sigmatropic reactions involve the movement of a sigma-bond with the simultaneous rearrangement of the π-system.
Two examples illustrate this:
The [1,5] shift of hydrogen in a 1,3-pentadiene system:
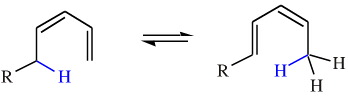
The [3,3] Cope rearrangement:
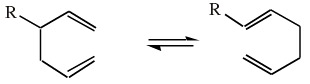
Note that the generic "R" functions have been added so that the product can be distinguished from the starting materials.
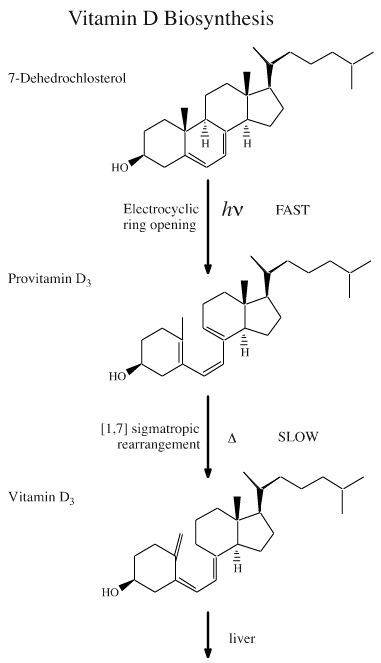
The biosynthesis of vitamin D has photochemical step, and the reaction takes place in skin cells.
A 1,3-cyclohexadiene system associated with the b ring of a steroid undergoes a photoactivated, retroelectrocyclisation to give 1,3,5-hexatriene system. (The thermal reaction is not allowed because it would be necessary for form a six membered ring with a trans-alkene, a sterically impossible structure.)
1,3,5-hexatriene system then undergoes a thermal [1,7] sigmatropic rearrangement to give vitamin D3. This is further processed in the liver.
Group Transfer Reactions
Group transfer reactions are a special class of complexation or fragmentation process. The are best exemplified by the ene reaction between a propene and ethene to give a 1-pentene.

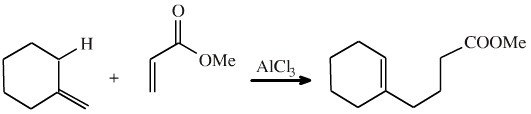
An example of an ene reaction is seen with the reaction of methylenecyclohexene and methyl acrylate. (An aluminium chloride catalyst is required to "activate" the enone system.)
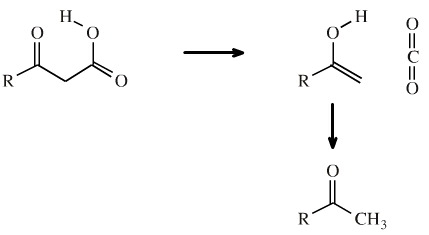
A common reaction in organic synthesis is the acid catalysed decarboxylation of a β-ketoester. The ester is hydrolysed to the β-ketoacid by the aqueous acid this rapidly loses carbon dioxide.
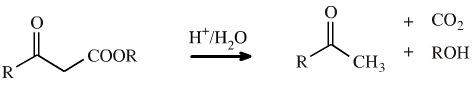
The reaction proceeds as a retroene reaction to give carbon dioxide and an enol system. The loss of CO2 drives the reaction to the right hand side. The enol rapidly tautomerises to the methyl ketone:
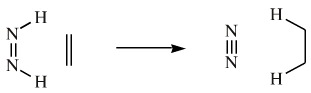
Diimide is used as a reducing agent, it adds H2 to C=C and N=N bonds and leaves other functions untouched:
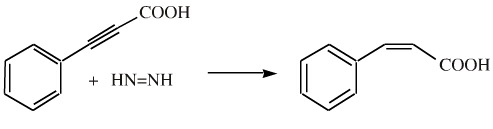
For example, 3-phenylpropynoic acid is reduced to cis-3-phenylpropenic acid (cis-cinnamic acid) by diimide:
Stereochemistry
Pericyclic reactions, because they are concerted, show extraordinary stereoselectivity, a topic outside the scope of the current iteration of this web book. The reader is advised to refer to Fleming, here.
More... and In More Detail
Have a look at Henry Rzepa's (of Imperial College) excellent web course:
- Home Page
- Historical
- Definitions
- Categories
- Theory
- Rules
- Electrocyclic
- Cycloaddition
- Sigmatropic
- Problems 1-10
- Problems 11-20
- Problems 21-30
- Problems 31-40
Tim Wallace, from the Universiry of Manchester, has three web courses:
|
|
| Addition Synthlet | Why Reactions Occur |
© Mark R. Leach 1999 –
Queries, Suggestions, Bugs, Errors, Typos...
If you have any:
Queries
Comments
Suggestions
Suggestions for links
Bug, typo or grammatical error reports about this page,please contact Mark R. Leach, the author, using mark@meta-synthesis.com
This free, open access web book is an ongoing project and your input is appreciated.