Periodic Table |
 |
 |
 |
 |
 |
 |
Chemogenesis Paper
This page presents the chemogenesis analysis as an academic paper. The language is more formal and there are fewer graphics, but this approach may be prefered by some readers.
Combinatorial Studies of the Main Group Elemental Hydrides and the Discovery of New Structure within Reaction Chemistry Space
Mark R. Leach
Division of Chemistry, School of Sciences, University of Salford, Gr. Manchester, M5 4WT, UK
Reaction chemistry space is explored by creating and combinatorially manipulating arrays of structurally simple chemical species derived from the first 36 s- and p-block (main group) elements of the periodic table. By rigorously exploring periodicity, previously unrecognised structure is exposed. Linear structural traits are found to be associated with more than 100 congeneric arrays and linear reactivity behaviours are often observed between interacting arrays. The regions of reaction chemistry space with innate, quantitative structure-reactivity behaviour are identified. The principle types of chemical reaction behaviour (Lewis acid/base, radical, diradical and redox) are seen to emerge from the periodic table. The analysis reveals how discontinuities in congeneric behaviour develop and it explains the anomalous organic chemistry of the fluoride ion.
Although many chemical reactions were known in the nineteenth century, an understanding of chemical reactivity had to wait until Bohr devised a plausible model of electronic structure in 1913 (1). Three years later Lewis produced his seminal paper (2,3), which introduced the idea of the two-electron covalent bond and established the rules for counting electrons in molecular structures and during chemical reactions which are still in use today. Electron accountancy and the concept of high-energy reactive intermediates led to mechanistic theory and the introduction of curly arrows to indicate electron flow. In the middle years of the twentieth century, much organic reaction chemistry was elucidated in terms of ionic and radical reaction mechanisms, with Robinson and Ingold leading the way (4,5). The tradition was continued by Woodward and Hoffmann, who explained an apparently diverse collection of organic reactions that involve p-systems in terms of pericyclic reactivity using frontier molecular orbital (FMO) theory (6,7).
Chemical reactivity amongst structurally simple chemical species was re-examined in the 1960s by Pearson, who developed the idea that ionic, polar and much non-polar reaction chemistry could be considered in terms of interacting Lewis acids and Lewis bases (8,9). A Lewis acid is an electron-pair acceptor and a Lewis base is a species with an available pair of electrons. A Lewis acid interacts with a Lewis base to give an acceptor—donor complex held together with a [Lewis] two-electron chemical bond (Box 1). Klopman (7,10) gave these ideas a modern footing by invoking a combination of coulombic (electrostatic) attraction and FMO theory. In the Klopman formulation there is a simultaneous +/– (charge—charge) attraction and the Lewis acid’s lowest unoccupied molecular orbital (LUMO) interacts with the Lewis base’s highest occupied molecular orbital (HOMO) to give a bonding molecular orbital (MO). The resultant Lewis acid/base complex may exhibit ionic (in Klopman’s parlance, charge-controlled) bonding, covalent (FMO-controlled) bonding or polar—covalent bonding (a mixture of charge and FMO contributions). A similar logic can be applied to free-radical species that have one unpaired electron in a singly occupied molecular orbital (SOMO).
Pearson attempted to rationalise chemical behaviour by classifying some 100 representative Lewis acids and Lewis bases as hard, borderline or soft. Within this schema, reaction chemistry behaviour was deemed to proceed according to the hard-soft acid-base (HSAB) principle: "hard [Lewis] acids prefer to bind to hard [Lewis] bases and soft [Lewis] acids prefer soft [Lewis] bases". The Pearson approach is highly successful when comparing pairs of species: the sodium ion Na+ is harder than the silver ion Ag+; alkoxide ions, RO–, are harder than thioanions, RS–; copper(II) ion, Cu2+, is harder than copper(I) ion, Cu+; the nitrogen anion end of the ambidentate cyanide ion, CN–, is harder than the carbon anion end, NC–.
However, the HSAB analysis is flawed because (1) no physical parameter correlates with hardness over Pearson's chosen set of species, (2) chemical interesting hard-soft interactions are not discussed and (3) the one-dimensional hard-borderline-soft continuum blurs much of the rich linear (predictable) behaviour which can be found.
An alternative system, which avoids and explains the pitfalls of the Pearson approach and which exposes previously unrecognised structure within main group chemistry, arises naturally from the new analysis presented here.
The Main Group Elemental Hydrides and Congeneric Arrays
Two chemical species are said to be isoelectronic when they have the same arrangement of electrons. It follows that isoelectronic species have the same Lewis structure, the same FMO topology and, often, mutually predictable reaction chemistry. When an array of species has a regularly changing electronic structure and a regularly changing reaction chemistry over the array, the members of the array can be said to be congeneric (11) or "of the same family" (12).
Congeneric arrays can be discovered and studied by generating normalised arrays of simple chemical species, examining the arrays for isostructural sub-arrays and then exploring these isostructural sub-arrays for linear behaviour. The master set of species upon which the analysis is performed consists of the first 36 main group elements, hydrogen to barium (i.e. only elements from the s- and p-blocks of the periodic table, groups 1, 2 and 13 to 18).
In their elemental state, the main group elements are highly diverse, consisting of metals, network-covalent metalloids and molecular non-metals. Allotropes abound. However, the structure and reactivity of the chosen set can be normalised by the saturation bonding of each main group element with a common bonding partner. There are several candidates, including hydrogen, fluorine, oxygen and lithium to give the corresponding main group hydrides, fluorides, oxides and lithides. By far the richest reaction chemistry emerges from hydrogen and the 36 corresponding main group elemental hydrides, H2 to BaH2.
All the main group elements form the stoichiometric hydride characteristic of their position in the periodic table in that they all exhibit the lower, common oxidation state or valency. The hydride of hydrogen is H2, the hydride of oxygen is H2O, the hydride of neon is Ne, etc. To further normalise the set to enable computer modelling experiments, all the species are deemed to be in the gas phase and molecular. Thus, lithium hydride is considered as the diatomic LiH(g) and not the ionic solid LiH(s).
The elemental hydrides can be chemically probed by adding or subtracting reactive hydrogen species: the proton (or hydrogen cation) H+, the hydride ion (or hydrogen anion) H–, and the hydrogen radical or (hydrogen atom) H. Five hydrogen-probe experiments can be upon the gas phase main group elemental hydrides (Figure 1).
The main group element to hydrogen bond lengths: X—H, [X—H]+ and [X—H]–, are a subtle and versatile indicator of reactivity. In specific circumstances (discussed below), the 'to-hydrogen' bond length' parameter correlates with the selectivity of the conjugate reactive intermediate: X– (anion), X+ (cation), X: (neutral Lewis base), X8(neutral Lewis acid) and X. (radical), for a bonding partner. X-to-hydrogen bond lengths can be determined to the necessary precision by ab initio calculation at the 3-21G* level.
Protons, hydride ions and hydrogen radicals make excellent structural and reactivity probes because they are electronically simple moieties of low mass, yet they act as surrogates for heavier species. The elemental hydrides and the compounds, atomic and molecular ions, radicals, etc., formed by the five hydrogen-probe experiments (Figure 1) are all simple species with uncontroversial structures and well known reaction chemistry. The low mass of the hydrogen-probe species means that they readily quantum tunnel between species (states) so that hydrogen transfer reactions (involving H+, H– and H.) form thermodynamic products because activation energy (kinetic) energy effects are minimal. The equilibrium position of the proton transfer, expressed in terms of the Brønsted acid’s Ka or pKa, gives information about both the Brønsted acid and about the conjugate Brønsted (and Lewis) base. Many pKa values and compilations of values appear in the literature (13,14,15,16).
The suitability of hydrogen as the common bonding element is enhanced by the problems associated with the other candidates (F, O and Li). The highly electronegative nature of fluorine means that the heavier p-block elements form higher oxidation state compounds, such as SF6, IF7, XeF6, etc. Oxygen is prone to give higher oxidation state compounds, but of more importance is that many oxides are refractory materials, which invalidates any molecular and in the gas phase assumptions. The chemistry of lithium is strongly influenced its small radius; Li does not even alloy with the other group 1 metals (17).
The Five Hydrogen-Probe Experiments
The main group elemental hydrides and the products of the five hydrogen-probe experiments are shown in Figure 1. Within the elemental hydrides and five hydrogen-probe experiments are 25 sub-arrays: eight of the sub-arrays are 1 x 1 dots, four are 4 x 1 series, six are 5 x 1 series and five are 4 x 4 planars. (The group 1 and 2 metal cation series appear twice, but are only counted once.) The series and planar sub-arrays are congeneric because the regularly changing structure correlates with linear reactivity behaviour over the arrays. While this statement is meaningless for 1 x 1 (and 1 x 2) arrays, the logic can be used to explore the reaction chemistry along the 4 x 1 and 5 x 1 congeneric series and over the 4 x 4 congeneric planars.
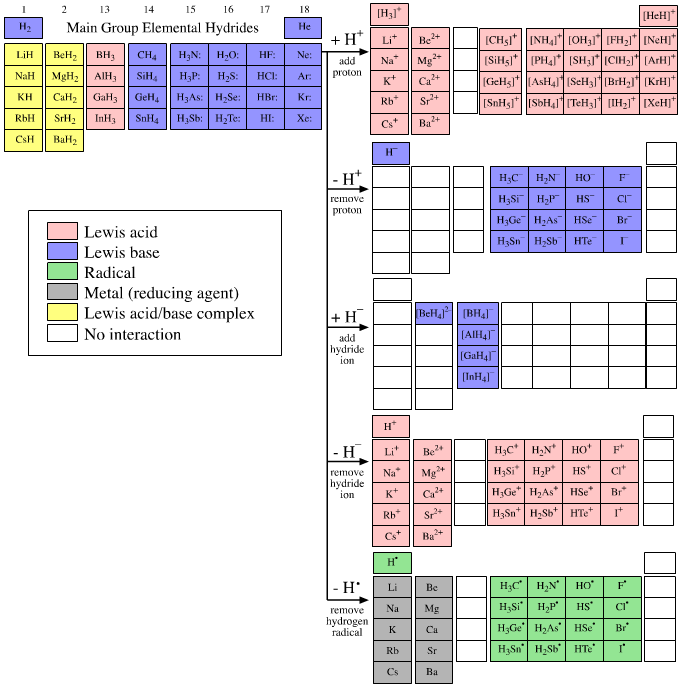
click on graphic for more info
Figure 1. The five hydrogen-probe experiments. Reactive hydrogen species, H+, H–, and H. are added and removed from the main group elemental hydrides. Not all interaction combinations are possible and many blank (null) interactions occur, for example Lewis acid plus Lewis acid. Reaction chemistry can also be more involved than simple complexation: the group 1 and 2 (saline) hydrides react with H+ to give the metal cation plus H2 and not a protonated complex of the type [LiH2]+. The group 13 hydrides dimerise, 2BH3 -> B2H6. Some of the equilibrium positions lie so far to the left that, for practical purposes, the reaction only occurs in the opposite direction to that indicated by the arrow, although this in no way invalidates the analysis. For example, a hydride ion cannot be generated by abstracting a proton from H2, because thereis no [Brønsted] base strong enough; however, a proton plus a hydride ion rapidly gives dihydrogen, H+ + H– -> H2.
Several tools can be used to correlate structure and reactivity and expose congeneric behaviour. These include: Lewis theory (Lewis electronic structure), FMO theory (HOMO, LUMO and SOMO topology), bond length (ab initio calculation), pKa (aqueous proton affinity with respect to water), gas phase proton affinity, electronegativity (elemental desire for electrons, x, revised Pauling), percentage ionic character (the Pauling equation = 16|xa-xb| + 3.5(xa-xb)2), ionic radius, reaction chemistry (rate data and the mapping of chemical behaviour onto Lewis and FMO theory).
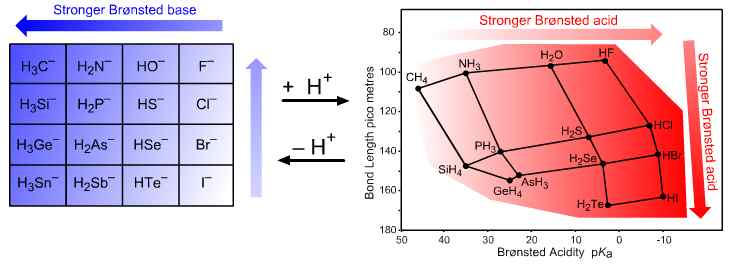
Figure 2. p-Block Brønsted acids and conjugate bases. The interaction of H+ with the group 14 to 17 anionic Lewis bases and their conjugate Brønsted acids shows planars of congeneric behaviour with respect to "to hydrogen" bond length and pKa. These two planars lie diagonally across the p-block and normal to each other. Brønsted acidity increases from CH4 to HI and bond length increases diagonally from HF towards SnH4. If the Pearson notion of hardness is equated with short conjugate acid bond length, F– is the hardest Lewis base and H3Sn– is the softest. When pKa values of the conjugate Brønsted acids are plotted against the corresponding bond length, the p-block elemental hydrides appear in their normal positions vis-à-vis the periodic table.
Congeneric Array Interactions
Each of the 25 congeneric sub-arrays shows distinct reaction chemistry behaviour. Ten of the congeneric arrays are Lewis bases, nine are Lewis acids, two are radicals and two are metallic. Congeneric behaviour can be readily probed amongst Lewis acid and Lewis base arrays (dots, series and planars) by examining the resultant complexes. The usual rules of array algebra are observed:
(1 x 1) x (1 x 1) → (1 x 1) dot x dot → dot
(5 x 1) x (1 x 1) → (5 x 1) series x dot → series
(4 x 4) x (1 x 1) → (4 x 4) planar x dot → planar
(5 x 1) x (4 x 1) → (5 x 4) series x series → planar
(5 x 1) x (4 x 4) → (5 x 4 x 4) series x planar → volume
The group I metal cation Lewis acid congeneric series Li+ to Cs+, a 5 x 1 array, interacts the hydride ion Lewis base, H–, a congeneric dot, to give a congeneric series of Lewis acid/base complexes, LiH to CsH, a (5 x 1) array. Regular patterns of structure and behaviour are found over both the Li+ to Cs+ and LiH to CsH series in terms of ionic radius, electronegativity, percentage ionic character, bond length, etc.
Likewise, the 5 x 1 Li+ to Cs+ congeneric series can be complexed with the 4 x 4 group 14 to 17 anionic Lewis base congeneric planar. The result is a 5 x 4 x 4 congeneric volume of chemical species (Figure 3).
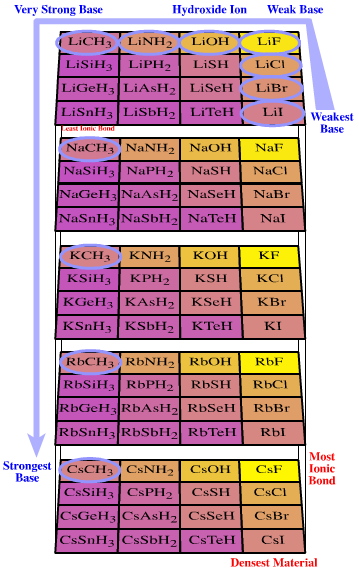
Figure 3. Congeneric volume. The 5 x 4 x 4 congeneric volume is formed when the 5 x 1 Li+ to Cs+ congeneric series of Lewis acids interacts with the 4 x 4 anonic Lobe-HOMO Lewis bases congeneric planar. The congeneric volume shows linear trends with respect to percentage ionic character and metal to heteroatom bond length. The object has various properties. Brønsted base proton-abstraction behaviour sweeps from LiI (which is Brønsted neutral) to LiF (F– is a weak Brønsted base), through LiOH (HO– is a strong Brønsted base), LiNH2 and LiCH3 (two ‘super bases’ of synthetic chemistry), to CsCH3, which this analysis predicts is the strongest proton-abstracting Brønsted base of the set, although the species is unknown. Caesium iodide is the highest density material and caesium fluoride has the most ionic bond.
Ligand Replacement
Isoelectronic, isostructural and isoreactive congeneric series can also be found amongst ligand replacement series, in which a defined atomic centre has its ligands changed in a manner that gives a regular change in electronic structure. This can occur in two ways, either the congeneric ligands can be exchanged with members of a congeneric series en masse, such as:
BF3 BCl3 BBr3 BI3
or a multiligated centre can have its ligands exchanged one at a time, such as:
[NH4]+ [RNH3]+ [R2NH2]+ [R3NH]+ [R4N]+
H3C– CH3H2C– (CH3)2HC– (CH3)3C–
Complementary array interaction chemistry can be explored amongst ligand replacement congeneric series. The 5 x 1 Li+ to Cs+ Lewis acid congeneric series can be arranged against the 4 x 1 complex anion Lewis base series [AlF4]–, [AlCl4]–, [AlBr4]–, [AlI4]– to give a 5 x 4 congeneric planar bounded by the ionic salts Li[AlF4], Li[AlI4], Cs[AlF4], Cs[AlI4].
Likewise, the 5 x 1 Li+ to Cs+ Lewis acid congeneric series can be arranged against the 4 x 1 methyl carbanion to tertiary butyl carbanion series H3C–, CH3H2C–, (CH3)2HC–, (CH3)3C– to give the corresponding alkyl/metal complexes of the type LiCH3and CsC(CH3)3.
Using congeneric array interaction logic, some 100 congeneric series, planars and volumes have been identified and are available in electronic form (20).
It is general that the comparative prediction of reaction behaviour can only be quantitative when the species are congeneric. It follows that the Pearson notion of hardness can only have quantifiable meaning with respect to members of a congeneric series. When we say that the lithium cation Li+ is softer than Na+, K+, Rb+ and Cs+, the term soft is being used as a proxy for those linear parameters (ionic radius, bond length, etc.), which map to linear structure and reactivity behaviour in congeneric complexes of these ions. On the other hand, the Pearson designation of the proton H+, the lithium cation Li+, the ammonium ion [NH4]+ and the iron(III) ion Fe3+ as being members of a set of hard Lewis acids is a different situation entirely. These species do not have regular Lewis or FMO electronic structural trends and they do not have mutually predictable reaction chemistry.
Reaction Chemistries
Congeneric dots, series, planars and volumes can be arranged, analysed and defined with respect to their reaction chemistry. Five general behaviours emerge: Lewis acid/base, radical, diradical, redox, and photo (Figure 4). This conclusion can also be reached by analysis of the various possible electron combinations available at the Lewis/FMO level of theory.
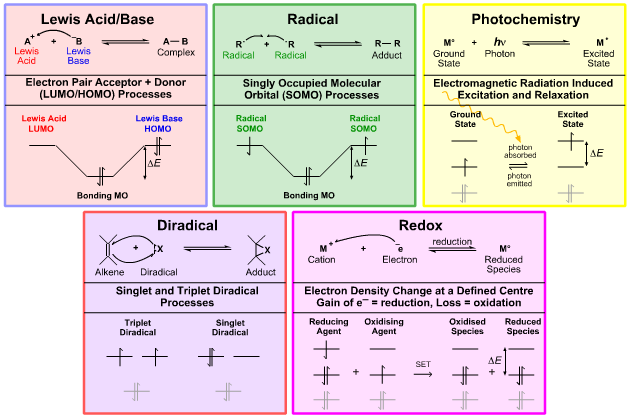
Figure 4. FMO analysis of the five reaction chemistries. The five reaction chemistries can be described in terms of Lewis theory curly arrows and frontier molecular orbital interactions. All reaction chemical behaviour that can be modelled at the Lewis or FMO levels can be mapped to one or more of these reaction chemistries.
This set of behaviours is comprehensive in that any reaction chemistry that can be modeled at the Lewis or FMO levels can be mapped to one or more of these five types. The hydride ion H– is a Lewis base and a reducing agent. The proton H+ is a Lewis acid and an oxidising agent. Dioxygen O2is a diradical and an oxidising agent. The benzene radical cation [C6H6]+• has both radical and Lewis acid properties. Photochemical and redox methods are used to produce radicals. Etc.
The richest—and the most widespread—behaviour is found amongst Lewis acid/base reaction chemistry. Complexes and complexation, anions and cations, lone pairs, ligands, counter ions, nucleophilic and electrophilic substitution and addition, Brønsted acidity, heterolytic fragmentation, ion paring, π-complexes, π/π interactions including Diels-Alder cycloaddition, ionic rearrangements, most redox chemistry and nearly all aqueous reaction chemistry can all be described in terms of interacting Lewis acids and Lewis bases. However, order can be brought to this miscellany of species by revisiting the hydrogen probe experiments (Figure 1).
The Lewis acids and Lewis bases generated by the hydrogen probe experiments, with the addition of conjugated π-systems and transition metal species, can be classified by their FMO topology. Six structurally distinct types of Lewis acid and four distinct types of Lewis base and can be recognised:
- The proton Lewis
acid H+ is a low-mass point-positive charge with a spherical
1s LUMO. The proton is the agent of Brønsted acidity
and is a unique Lewis acid.
- s-LUMO Lewis acids, cations of the group I and II metals (Li+, etc., and Be2+, etc.).
Species have a net positive charge and a spherical 2s, 3s , 4s, 5s or 6s LUMO layer superimposed upon a
full-shell electronic structure. Species are commonly act as spectator
counter ions to anionic Lewis bases.
- Onium ion Lewis acids ([H3O]+, [(CH3)4N]+,
etc.), have a central atom hypersaturated with H+ or R+ ligands. Species either behave as H+ (Brønsted acid)
donors or alkylating agents (R+ donors), depending upon
the ligand. Non-basic and/or non-nucleophilic spectator counter ions are required.
- Lobe-LUMO Lewis acids either have a vacant p-orbital (F3B•,
H3C+, etc.), or have a bond with
a mixed-in p-orbital component, which makes it susceptible
to nucleophilic attack (methyl iodide, H3C—I, carbonyl functions, alpha,beta-unsaturated carbonyl functions, epoxides,
etc.). Cationic Lobe-LUMO Lewis acids, such as Cl+, are
amongst electrophilic species. Cationic species require a non-nucleophilic
counter ion such as [AlCl4]–.
- π-LUMO
Lewis acids (allyl cation, benzyl cation, etc.) have a cationic or electron-poor p-system delocalised over two or more p-orbitals
that partakes in multicentre interactions. For example, when the allyl
cation [C3H5]+ interacts with Pd2+ to give an allyl
palladium complex, it is behaving as a p-LUMO Lewis acid. However,
it should be noted that when the allyl cation reacts with a nucleophile,
for example H– to give propene, at a terminal carbon
(and a single p-orbital), it is behaving as a Lobe-LUMO Lewis
acid.
- Heavy metal Lewis acids (Cu2+, Pd2+,
etc.) include all transition, post-transition, lanthanide and actinide
metal ions and bulk metals. Heavy metal Lewis acids have partially
filled and vacant d and f orbitals (which may hybridize) available for both bonding and back-bonding. Pearson recognised that species become harder with increasing oxidation state (Fe3+harder than Fe2+) and softer down a group (Ni2+is harder than Pd2+, which is harder than Pt2+).
- s-HOMO Lewis
bases (H– and H2) have a spherical
or ovoid highest occupied molecular orbital. Hydride ion is a very
powerful proton abstractor and hydrogen a very weak proton abstractor.
Both species are reducing agents.
- Complex anion Lewis
bases ([BH4]–, [AlF4]–, etc.) are full-shell net-negative charge species with anionic ligands
(H– to X–) that surround an electropositive
atomic centre. With a suitable counter ion, usually Li+,
Na+ or K+, reagents act as ligand-donor species.
- Lobe-HOMO Lewis
bases are classic lone pair of electron species: HO–,
H2O:, Et2O:, H2N–,
H3N:, H3C–,
carbanions, halogen anions, alpha-effect bases such as H2NOH
and HOO–, ambidentate bases such as CN– and NO2–, bidentate bases like
dimethyl ethylene glycol and tetramethylethylenediamine and polydentate
bases like crown ethers and cryptands.
- π-HOMO
Lewis bases (ethylene, benzene, allyl anion, cyclopentadienyl anion,
benzyl anion, etc.) have an anionic or electron-rich p-system delocalised
over two or more p-orbitals and partake in multicentre interactions.
For example, when the cyclopentadienyl anion [C5H5]– reacts with Fe2+ to give ferrocene,
it is behaving as a π-HOMO
Lewis base. However, when the cyclopentadienyl anion is protonated
to cyclopentadiene at a single p-orbital, it is behaving as
a Lobe-HOMO Lewis base.
The six types of Lewis acid and four types of Lewis base interact to give a matrix with 24 types of Lewis acid/base complex (21) and many properties (Figure 5).
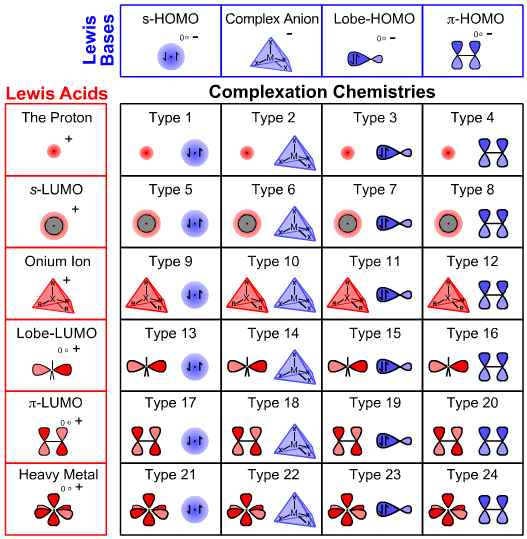
Figure 5. The Lewis acid/base interaction matrix. The six types of Lewis acid and four types of Lewis base interact to give 24 distinct types of Lewis acid/base complex and associated complexation chemistry. The matrix has many properties, for example: most basic reagents, such as sodium hydroxide, calcium carbonate and methyl lithium, are Type 7 complexes; π/π interactions—including Diels-Alder cycloaddition chemistry—is associated with Type 20 complexation; classical inorganic coordination chemistry is associated with Type 23 complexation and Type 24 complexation includes all π/transition metal chemistry. Congeneric series, planars and volumes are always found within cells and never straddling cells. The extensive properties of the matrix are discussed in ref. 21.
Non-linear Behaviour and The Emergence of Organic Chemistry
Linear structural trends that run over congeneric arrays do not always map to linear reaction behaviour. When the 4 x 1 carbenium ion Lewis acid congeneric series [H3C+, CH3H2C+, (CH3)2HC+, (CH3)3C+] is arranged against the 4 x 1 anionic Lewis base congeneric series (H3C–, H2N–, HO–, F–), the resultant 4 x 4 array of species is linear with respect to bond length and percentage ionic character, but is non-linear with respect to our common experience of the reaction chemistry (Figure 6).
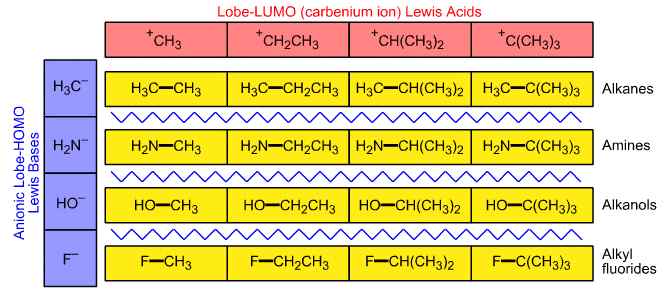
Figure 6. The emergence of organic chemistry. When the carbenium ion Lewis acid congeneric series H3C+, CH3H2C+, (CH3)2HC+, (CH3)3C+ complexes with the period 2 anionic Lewis base congeneric series H3C–, H2N–, HO–, F–, the resultant array of species consists of alkanes, amines, alkanols and alkyl fluorides. The 4 x 4 array is non-linear with respect to reaction chemistry behaviour bifurcates (forks).
The products of the carbenium ion/period 2 anion interaction are alkanes, amines, alkanols and alkyl fluorides and the formation of these chemically distinct species has marked the place in reaction chemistry space where organic chemistry breaks away from main group chemistry and assumes its own distinct identity. The reason why this particular planar bifurcates (forks) is twofold. Firstly, the alkanes are not congeneric with respect to the corresponding amines and alkanols because the alkanes do not possess a functional group with a lone pair of electrons. Secondly, aqueous solubility and the multiple reaction pathways that become available over the pH range from concentrated hydrochloric acid to saturated sodium hydroxide solution dominate our common experience of these substances.
Bases, Nucleophiles and Organic Chemistry
Fluoride, F–, is congeneric with the other halogen anions, both with respect to the proton H+ and the Group I metals Li+ to Cs+. But the fluoride ion is not congeneric with chloride, bromide and iodide when the Lewis acid is a carbenium ion, H3C+ or R3C+. The result that the fluoride ion appears to have an anomalous reaction chemistry. To explore this system, consider the proton H+ and the methyl cation (or carbenium ion) H3C+ Lewis acid complexing with H3C–, H2N–, HO–, F– and with F–, Cl–, Br–, I– (Figure 7). Within this restricted system we can resurrect the Pearson notion of hardness and, first, ask the question, "Which end of the H3C–, H2N–, HO–, F– congeneric series is harder?"
The methyl anion H3C– (as methyl lithium, H3CLi) is a super-base, and on the Ka scale the methyl anion proton abstracts some 43 orders of magnitude more aggressively than the fluoride ion F–. (Conjugate acid data: CH4Ka = 1–46, HF Ka = 1.8–4).
However, the fluoride ion has a shorter F– to H+ bond length as its conjugate Brønsted acid than does the methyl anion (HF = 94 pm, CH4 = 108 pm), which makes the fluoride ion appear harder than the carbanion.
The hardness of the H3C– to F– congeneric series is defined with respect to bond length. A short ‘to H+’ or a short ‘to H3C+’ bond length equates with hard and a long ‘to hydrogen’ or ‘to carbon’ bond length equates with soft. (Using this bond-length logic, the methyl cation H3C+ is of the same hardness as the methyl anion H3C–.) The fluoride ion F– has a shorter to H3C+ bond length than H3C– (F—CH3 = 140 pm, H3C—CH3 = 154 pm), so F– is harder than H3C–. However, the chloride, bromide and iodide anions have longer to H3C+ bond lengths than H3C– (Cl—CH3 = 178 pm, Br—CH3 = 195 pm and I—CH3 = 218 pm), so the heavier halogen anions are softer Lewis bases than H3C–. These structural trends are mirrored in the SN1, SN2, E1 and E2 reaction chemistry at carbon centres. Fluoride ion’s behaviour is linear with respect to its congeneric siblings HO–, H2N– and H3C–, but not with respect to Cl–, Br– and I–. The fluoride ion F– is moderately nucleophilic (F– < HO– < H2N– < H3C–), yet seldom behaves as a nucleofugal (Nfg–) Lewis base leaving group (F– > HO– > H2N– > H3C–). On the other hand, chloride, bromide and iodide are generally rather poor nucleophiles at carbon centres, but they make excellent nucleofuges (Cl–< Br–< I–).
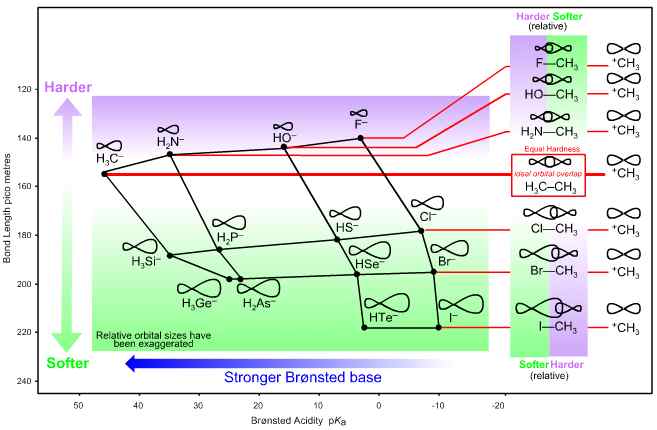
Figure 7. Lobe-HOMO Lewis base hardness and bond length. The graph shows the ‘to carbenium ion’ bond length of the anionic Lobe-HOMO array plotted against pKa. The C—C bond length associated with ethane, formed when H3C+ complexes with –CH3 (a type 15 Lewis acid/base complexation), is of intermediate length, 154 pm. If the carbenium ion, H3C+, is deemed to be of the same hardness as the carbanion, —CH3 then "harder-softer" arguments reverse between F—CH3 (hard/soft) and Cl—CH3 (soft/hard). Therefore, F– cannot be congeneric with Cl–, Br– and I– with respect to H3C+.
Conclusion
Chemogenesis, the process of chemical structure and reactivity emerging from the periodic table of the elements as described here has several stages. First, combinatorial arrays of simple chemical species are generated, iso-structural sub-arrays identified and tested for iso-reactivity. Those arrays that are iso-structural and iso-reactive are deemed to be congeneric, a term that quantifies the nineteenth century idea of periodicity. Analysis at the Lewis and FMO levels shows that most arrays are populated by species recognised as Lewis acids, Lewis bases or Lewis acid/base complexes. Complimentary Lewis acid/base array interaction chemistry can be used to test and define congeneric behaviour. Starting from the main group hydrides, the combinatorial approach illuminates reaction chemistry space and exposes congeneric dots, series, planars and volumes (Figure 3), the five reaction chemistries (Figure 4) and the Lewis acid/base interaction matrix (Figure 5).
The analysis also clarifies the proton-abstracting and/or nucleophilic facets of behaviour associated with Lewis bases. On first inspection, it seems that nucleophilicity should anti-correlate with the pKa of the Lewis base’s conjugate Brønsted acid (i.e., a stronger base equals a weaker nucleophile, but it does not. In the 1960s, it was hoped that Pearson's hardness parameter would provide the necessary correlation, but the chosen set of species was too diverse. The analysis presented here shows that nucleophilic and proton-abstracting behaviour do correlate with structure when the measure is bond length of the generated Lewis acid/base complex with two provisos. First, the complexing Lewis acid must be standardised: behaviour towards H+ is different to behaviour towards H3C+. Second, the Lewis base species must be members of a congeneric series. However, the vast majority of chemical species are isoelectronically unique and they do not have congeneric siblings. Quantitative structure activity relationships (QSAR) are rare in reaction chemistry space.
Methods
All bond lengths are determined by calculation using Spartan Pro (WaveFunction Inc.) Calculations are performed at the restricted Hartree-Fock 3-21G* and 6-31G** levels and at the density functional SVWN/DN* and pBP/DN** levels. A particular congeneric series, planar or volume is always modelled using the same level of theory.
|
Box 1 The Lewis and Brønsted Theories of Acidity The Lowey—Brønsted theory has a (Brønsted) acid as a proton donor. The Lewis theory of acidity has a (Lewis) acid as an electron-pair acceptor. The Lewis model is more general than the Brønsted model, and the two theories can be reconciled by recognising that the proton, H+, is a unique and versatile Lewis acid and is the agent of Brønsted acidity. All Brønsted acids are proton/Lewis base complexes and the transfer of H+ between Lewis bases equates with Brønsted acidity. While a Brønsted acid is an H+ donor, the proton, H+, is a Lewis acid. All Lewis bases can be protonated and the ability to complex a proton defines a species as being both a Brønsted base and a Lewis base. Finally, any species able to complex with a Lewis base is a Lewis acid. Consider the reaction of hydrogen chloride with water: HCl + :OH2 → [H3O]+ + Cl– In the Brønsted description, HCl is the proton-donor (Brønsted) acid and water is the proton-accepting (Brønsted) base. On the right-hand side of the equation, the oxonium ion, [H3O]+, is the conjugate (Brønsted) acid and the chloride ion, Cl–, is the conjugate (Brønsted) base. The Lewis model considers chloride ion and water to be Lewis bases, which compete to complex with the proton Lewis acid: hydrogen chloride, a H+/Cl– complex, transfers H+ to water to give the [H3O]+ (or H+/:OH2) oxonium ion complex. In the Brønsted analysis all proton acceptors (bases) are standardised against the aqueous base :OH2. Therefore, the term Brønsted base refers to proton affinity with respect to water while the term Lewis base refers to the general propensity to complex with a Lewis acid. |
References
1. Bohr, N. On the constitution of atoms and molecules, Phil. Mag. 26, 476—501 (1913).
2. Lewis, G.N. The atom and the molecule. J. Am. Chem. Soc. 38, 762—785 (1916).
3. Lewis, G.N. Valence and Structure of Atoms and Molecules, The Chemical Catalogue Company, New York (1923).
4. Ingold, C.K. Structure and Mechanism in Organic Chemistry, Cornell University Press, New York (1953).
5. Brock, W.H., The Fontana History of Chemistry, 506—569, Fontana, London (1992).
6. Woodward, R.B. and Hoffmann, R. The Conservation of Orbital Symmetry, Verlag Chemie, GmbH, Weinheim/Bergstr. (1971).
7. Fleming, I. Frontier Orbitals and Organic Chemical Reactions, John Wiley and Sons, Chichester (1976).
8. Pearson, R.G. Hard and soft acids and bases. J. Am. Chem. Soc. 85, 3533—3539 (1963).
9. Pearson, R.G. Hard and Soft Acids and Bases, Dowden, Hutchinson & Ross Inc, Stroudsburg, PA (1973).
10. Klopman, G. Chemical reactivity and the concept of charge- and frontier- controlled reactions. J. Am. Chem. Soc. 90, 223—234 (1968).
11. Ho, T.-L. The hard soft acids bases (HSAB) principle and organic chemistry. Chem. Rev. 75, 1—20 (1975).
12. The Oxford English Dictionary, Clarendon Press, Oxford (1989).
13. Jolly, W.L. Modern Inorganic Chemistry, McGraw-Hill Book Company, New York (1984).
14. March, J., Advanced Organic Chemistry, 218—236, John Wiley & Sons, New York (1985).
15. Pearson, R.G. Ionization potentials and electron affinities in aqueous solution. J. Am. Chem. Soc. 108, 6109—6114 (1986).
16. Lowry, T.H. and Richardson, K.S., Mechanism and Theory in Organic Chemistry, 293—296, Harper-Row, New York (1987).
17. Lee, J.D. Concise Inorganic Chemistry, Blackwell Science, Oxford (1996).
18. Olah, G.A., Prakash, G.K.S. and Sommer, J. Superacids, John Wiley & Sons New York (1985).
19. Morris, D.F.C. Ionic radii and enthalpies of hydration of ions. Structure and Bonding 4, 63—82 (1968).
20. Leach, M.R. http://www.meta-synthesis.com
21. Leach, M.R. Lewis Acid/Base Reaction Chemistry, meta-synthesis, Brighton (1999).
|
|
| The Chemical Thesaurus | Electronegativity Paper |
© Mark R. Leach 1999 –
Queries, Suggestions, Bugs, Errors, Typos...
If you have any:
Queries
Comments
Suggestions
Suggestions for links
Bug, typo or grammatical error reports about this page,please contact Mark R. Leach, the author, using mark@meta-synthesis.com
This free, open access web book is an ongoing project and your input is appreciated.